Do you want to buy antibiotics online without prescription? https://buyantibiotics24h.net/ - This is pharmacy online for you!
Approaches to assessing drug safety in the discovery phase: highlights of the society for medicines research symposium
Copyright 2010 Prous Science, S.A.U. or its licensors. All rights reserved.
APPROACHES TO ASSESSING DRUG SAFETY INTHE DISCOVERY PHASE
HIGHLIGHTS OF THE SOCIETY FOR MEDICINESRESEARCH SYMPOSIUM HELD ON SEPTEMBER 24TH, 2009, AT THE NATIONAL HEART &LUNG INSTITUTE, KENSINGTON, LONDON, U.K. J. Allen1, P. Jeffrey2, R. Williams3 and A.J. Ratcliffe4
1AstraZeneca, Alderley Park, Macclesfield, SK10 4TG, UK; 2GlaxoSmithKline, Immunoinflammation Centre of Excellence for Drug Discovery,Gunnels Wood Road, Stevenage, SG1 2NY, UK; 3Cancer Research UK, P.O. Box 123, WC2A 3PX, UK; 4Cellzome Ltd., Chesterford ResearchPark, Little Chesterford, Cambridge CB10 1XL, UK
Professor Kevin Park (MRC Centre for Drug Safety Science,Department of Pharmacology, University of Liverpool, U.K.) deliv-
The Society for Medicines Research symposium, sponsored by
ered the opening lecture on molecular aspects of adverse drug reac-
Apredica, Gwathmey Preclinical Services, Gentronix and Cyprotex, was
tions from molecule to man. Professor Park suggested understand-
held at the National Heart and Lung Institute, Kensington, London,
ing adverse drug reactions at the molecular level, and linking them
U.K. The meeting, organized by Jack Allen, Phil Jeffrey and Andrew
to pharmacogenomics, offered a way of tailoring the medicine to the
Ratcliffe, focused on approaches to assessing drug safety in the discov-
individual patient to minimize unwanted adverse toxicity. To illus-
ery phase. Topics included molecular aspects of adverse drug reactions
trate the approach, warfarin therapy was given as an example. from molecule to man, the relationship of physicochemical properties
Warfarin produces anticoagulation by reducing the binding of coag-
to toxicity, reactive metabolites, inhibition of the cardiac sodium chan-
ulation factors to the vascular endothelium. At the mechanistic level,
nel, drug-induced mitochondrial dysfunction, high-throughput screen-
this is driven through inhibition of vitamin K epoxide reductase com-
ing for genotoxicity and carcinogenicity, and the use of zebrafish as a
plex (VKORC1), an enzyme that is a key operator in the post-transla-
model for hepatotoxicity and developmental toxicity.
tional γ-carboxylation of glutamic acid residues on the coagulation
Attrition in drug development is still cripplingly high, with toxicity
factors required for endothelial binding. Warfarin is administered as
the leading cause at all stages in the drug development pipeline. It
a racemate, with the more potent (S)-isomer undergoing metabo-
has been estimated that a 10% improvement in predicting failure
lism principally by cytochrome P450 CYP2C9. However, there are
before the initiation of expensive and time-consuming clinical trials
certain groups of patients which represent challenges to the dosing
could save upwards of $100 million in the costs associated with drug
and management of warfarin therapy (1-3). Patients with a common
development. Furthermore, since approximately 70% of all toxicity-
functionally defective CYP2C9, resulting in an inability to efficiently
related failures that occur preclinically are comprised of toxicologi-
eliminate (S)-warfarin from the systemic circulation through metab-
cal outcomes for which the preclinical models are predictive of
olism, run a higher risk of life-threatening bleeding. To compensate,
human toxicity, then the benefits of identifying and predicting safe-
a significantly lower maintenance dose regimen is required. There
ty liabilities earlier in the drug discovery and development process
are also patients with VKORC1 polymorphisms, which can result in
could be of enormous benefit and value.
either warfarin sensitivity, as in the case of CYP2C9 polymorphism,or warfarin resistance, in which patients are stratified to higher doserequirements. As a consequence of understanding in detail themechanism of a drug’s action, and associated genetic and environ-
Correspondence: Andrew J. Ratcliffe, Cellzome Ltd., Chesterford Research Park, Little Chesterford, Cambridge CB10 1XL, UK. E-mail: andrew.ratcliffe@cellzome.com.
mental factors that avoid adverse drug reactions, new regulatory
APPROACHES TO ASSESSING DRUG SAFETY IN THE DISCOVERY PHASE
labeling can be approved and subsequently used by physicians to
bioactive thiol is further compromised, leading to nonresponsive-
deliver improved healthcare. In the case of warfarin, new labeling
ness and a higher rate of adverse clinical events. In an acute coro-
was approved in 2007, in which genetic variations of CYP2C9 and
nary setting this can take the form of death from cardiovascular
VKORC1, along with other factors, should be considered when pre-
causes, myocardial infarction or stroke. In comparison to clopidogrel,
scribing the drug, although such tests should not delay initiating
the onset of action of prasugrel is rapid and more sustained, and it
is less prone to adverse events involving CYP2C19 polymorphism,given a lack of involvement of this CYP in its bioactive thiol genera-
In another example, the importance of mechanistically understand-
ing at the human level the conversion of prodrugs to their activeform, and placing in context the risk of adverse drug reactions, was
The hepatocyte is well tuned to sensing chemical stress caused by
demonstrated in comparing and contrasting the clinically approved
exposure to reactive metabolites. With respect to the molecular
receptor antagonists clopidogrel (1) and prasugrel (2) (4, 5).
mechanisms that come into play, a huge amount of fundamental
Although both drugs require biotransformation to generate active
information has been mapped out from a detailed understanding of
thiols that subsequently bind irreversibly to cystine residues in the
events caused by dosing of paracetamol (6). As part of the disposi-
P2Y ADP receptor on platelets, the pathways to such species differ
tion of paracetamol, a small proportion is converted to the reactive
metabolite N-acetyl-p-benzoquinoneimine (NAPQI), which isquenched by hepatic glutathione (GSH) or cysteine residues within
Through clever design, the ester group in prasugrel is hydrolyzed
KEAP1, a cytosolic protein that binds to the redox-sensitive tran-
using intestinal hydroxy esterases to the thiolactone (3), which is
scription factor Nrf2. Under normal physiological conditions, the role
subsequently metabolized primarily by CYP3A4 to its active thiol (4).
of KEAP1 is to maintain Nrf2 transcription in check through a protea-
In contrast, clopidogrel is transformed to its active thiol (5) via an
some-dependent degradation of the protein. This process is disrupt-
intermediate thiolactone (6) solely through CYP1A2-, CYP3A4-,
ed by the covalent binding of NAPQI, with liberation of Nrf2 and sub-
CYP2C9- and CYP2C19-dependent transformations.
sequent translocation to the nucleus, where it orchestrates an
The drug disposition of clopidogrel is further complicated by the fact
antioxidant response through the activation of cell defense genes,
that only approximately 15% of the drug is metabolized by the CYP
such as glutathione transferases, NAD(P)H quinine oxidoreductase,
pathway to its bioactive thiol form, with the majority suffering
heme oxygenase and glucuronyltransferase. Unfortunately, at a high
hydrolysis to an inactive acid derivative (7). As a result, patients often
(over) dose of paracetamol the extent of NAPQI formation seriously
receive a relatively low exposure of the active form of the drug, which
depletes hepatic GSH levels and overwhelms the antioxidant line of
can lead by its very nature to a variable onset of action and response.
defense, resulting in the covalent modification of critical proteins,
This is further heightened in patients carrying a CYP2C19 reduced
such as γ-glutamylcysteine synthetase, glyceraldehyde-3-phos-
functional allele, in which their ability to metabolically generate the
phate dehydrogenase (GAPDH) and Ca2+/Mg2+-ATPase, which are
Figure 1. Biotransformation of clopidogrel (1) and prasugrel (2) to active thiols.
THOMSON REUTERS – Drugs of the Future 2010, 35(1)
APPROACHES TO ASSESSING DRUG SAFETY IN THE DISCOVERY PHASE
key in maintaining mitochondrial function and Ca2+ homeostasis.
To fill this void, GSK initiated an internal program aimed at defining
The impact of such an insult on the cell can lead ultimately to cell
certain guidelines. In addition, it was hoped that such an output
death, either by apoptosis or necrosis. Of great interest is that both
could help address the high proportion of compounds failing to
forms of cell death release specific signature proteins that can be
progress within the organization due to unacceptable hepatoxicity.
monitored in plasma, namely high-mobility group box protein 1
To this end, a database was established consisting of 200 hepato-
(HMG-1) from necrotic cells and fragments of cytokeratin-18 (CK-18)
toxic and 200 nonhepatotoxic known drugs. Criteria for entry into
from apoptotic cells. From animal studies, increased doses of para-
the hepatotoxic class of drug consisted of at least 50 reports of
cetamol, up to 550 mg/kg, have been correlated with increased
hepatotoxicity or at least 3 reports of life-threatening hepatotoxicity,
plasma levels of these signature proteins, although each one has a
warnings or precautions in label, including liver enzyme monitoring.
unique dose–response profile. Above doses of 550 mg/kg and up to
In contrast, no reports of hepatotoxicity or only one report of “mild”
1000 mg/kg, the levels of CK-18 fragments captured dramatically
hepatoxicity constituted the nonhepatotoxic class of drugs.
fall, while in contrast, levels of HMG-1 continue to rise. From the
Initially, two methodologies, often used in lead optimization, were
unraveling of the molecular mechanisms of paracetamol toxicity,
used to assess the propensity for reactive metabolite formation. The
HMG-1 and CK-18 fragments appear as potential biomarkers for cel-
first centered on GSH trapping, where production of GSH conjugates
lular necrosis and apoptosis, respectively. Such relationships have
from microsomal incubations was monitored via loss of pyroglutam-
found utility in the diagnosis and severity grading of a number of dis-
ic acid (m/z 129). Using a signal:noise ratio approach, the extent of
GSH conjugative production could be quantified (ratios: minor 1-10;
Hypersensitivity is a common adverse event with many drugs that
notable 10-100; and marked > 100). The second assay focused on
can limit their therapeutic use; examples include carbamazepine,
CYP time-dependent inhibition (TDI). The CYPs employed covered
sulfamethoxazole, abacavir and nevirapine. There is a growing body
1A2, 2C9, 2C19, 2D6, 3A4DEF and 3A47BQ, and the extent of the
of evidence that reactive drug metabolites may play a key role in elic-
fold change with time allowed a ranking distribution of the
iting the allergic reaction, which is believed to center on activation of
drugs into subsets (fold shift: minor < 2; notable 2-5; and marked >
the T-lymphocyte system (9). Although a lymphocyte transformation
5). Results from the GSH trapping screen revealed that 80% of GSH
test (10) is available as an in vitro test to assess the propensity of a
adducts classified as marked adhered to the hepatotoxic class of
drug or its metabolite to cause hypersensitivity, further research has
drugs. In the case of marked TDI, this figure dropped to 70%.
revealed a genetic basis that may offer pharmacogenetic screening
However, common to both screens was a cluster of drugs displaying
as a means of de-risking patient drug hypersensitivity. In several
marked GSH/TDI effects but not exhibiting hepatotoxicity. Further
clinical studies of abacavir, a nucleoside reverse transcriptase
investigation of this cluster revealed that many of the drugs were
inhibitor designed to combat HIV, avoiding inclusion of patients with
administered as a low dose (< 10 mg) or intermittently or topically
the major histocompatibility complex (MHC) class I allele HLA-
applied. Revisiting the analysis and setting the drug dose threshold
B*5701 delivered a significantly reduced diagnosis of hypersensitivi-
to > 100 mg/day delivered an improved discrimination. Of those
ty (11). Such results represent a potentially important step in the clin-
drugs that exhibited hepatotoxicity in man, 96% and 82%, respec-
ical management of abacavir, given that patients presenting
tively, gave marked GSH adducts and TDI effects.
hypersensitivity manifest conditions of fever, rash, gastrointestinal
The availability of radiolabeled drugs allows a further assessment of
and respiratory symptoms that can become more severe, rapid and
the extent of drug protein adducts and the risk of toxicity, in partic-
life-threatening if discontinuation of the drug is not immediately
ular idiosyncratic toxicity (IDT), through bioactivation mechanisms.
instigated. The HLA-B*5701 genotype has also been advocated as a
Covalent binding measurements can be performed in liver microso-
high risk factor for the drug-induced liver injury caused by flu-
mal or hepatocyte preparations from both animals and humans. In
cloxacillin, and prescreening of patients, as in the case of abacavir,
the case of animals, the level of adducts to both liver and plasma
may provide enhanced patient compliance (12).
proteins can be determined in the intact animal after oral dosing.
Mr. Andy Harrell (GSK, U.K.) presented a current industry perspective
Seminal work by the Merck group (13) suggested 50 pmol drug
on reactive metabolites. Since their identification in the 1930s, there
eq/mg protein as a target upper limit, above which a compound
has been great interest in the area. During the last 30 years of the
would not be advanced into development unless other qualifying
20th century, the association of reactive metabolites with toxicity and
considerations were taken into account, such as a daily dose < 10 mg
withdrawal of certain classes of drugs, such as β-lactam antibiotics
or if the disease under treatment was life-threatening.
(anaphylaxis), nonsteroidal anti-inflammatory drugs (NSAIDs; idio-
As part of the GSK predictive toxicology initiative, 65 radiolabeled
syncratic hepatoxicity) and arylamines (carcinogenicity and agranulo-
drugs of known safety profiles were screened in a microsomal acti-
cytosis), began to emerge. Several hypotheses (hapten and critical
vation assay, in which residual radioactivity associated with unex-
protein) were proposed to support the link at a mechanistic level, in
tracted protein was measured. Analysis of the results suggested 200
which a central tenet focused on covalent binding of the reactive
pmol drug eq/mg of protein as an alert. In a second GSK study,
metabolite to proteins or DNA. As a consequence, during the last 10
analysis of ex vivo covalent binding data, easily generated from low-
years a multitude of nonradioactive reactive metabolite screens have
dose toxicology studies, suggested that > 30% unrecovered radioac-
become engrained within drug discovery in an attempt to identify
tivity at a concentration of > 1 pmol represented a further risk alert.
development candidates where risk from reactive metabolite genera-tion and toxicity is minimized. However, during the early period of this
Reference was also made to recently published work by researchers
paradigm shift to reactive metabolite screening, little scientific data
at Daiichi Sankyo (14), who screened 42 radiolabeled drugs of known
existed in the public domain to support decision-making.
safety profiles for covalent binding in human microsomes, human
THOMSON REUTERS – Drugs of the Future 2010, 35(1)
APPROACHES TO ASSESSING DRUG SAFETY IN THE DISCOVERY PHASE
hepatocytes and rat liver in vivo. They also concluded that 50 pmol
cology of the compound were removed from the analysis. Defined
drug eq/mg of protein was ineffective in grouping the safety cate-
lists of significant toxicology markers, generated from histopatholo-
gories, with drugs given a warming of IDT in the Physician Desk
gy, clinical pathology or clinical signs, were used to label the com-
Reference and Japanese drug labeling not exceeding this threshold,
pounds. Free drug exposure could be substituted for total drug
and 4 of the 12 so-called safe drugs exhibiting covalent binding in
exposure, providing plasma protein binding data were available.
human microsomes that exceeded 100 pmol drug eq/mg of protein.
Unfortunately, due to a lack of historical data, only 72% of the origi-
Indeed, the use of covalent binding alone in each of the three test
nal dataset met this criteria. However, by following a similar line of
systems failed to distinguish the safety categories. Only when clini-
data deconvolution, the appropriate free drug C
cal daily dose was taken into account did a significant distribution
determined to be 1 µM. Regardless of whether free or total drug Cmax
plot become clear, with the separation of drugs into safe, equivocal
was examined, analysis of the drug toxicity classification using a
wide range of physicochemical properties and descriptors revealed aconsistent link with both TPSA and CLogP. Calculation of toxicity
In concluding the talk, Mr. Harrell suggested there was a move away
odds ratios revealed an inter-relationship between these properties,
from blanket screening for reactive metabolites to a more bespoke
and by setting cut-offs for CLogP and TPSA of 3 and 75 A2, respec-
application of various screening models that could be applied at dif-
tively, identified compounds with CLogP > 3 and TPSA < 75 A2 as
ferent stages of a project. Risk assessment guidelines were now
being six times more likely to have an impactful toxic outcome than
available from such models, with several incorporating a clinical
compounds with CLogP < 3 and TPSA > 75 A2, whether based on
dose input that led to a higher level of predictivity. As an underlying
total or free drug exposure. Compounds exhibiting only a single risk
theme, increased dose appeared to be a clear driver for increased
factor gave a weak and inconsistent trend. The addition of a further
2 years of new IVT data reinforced the high CLogP (> 3)/low TPSA
Professor Julien Blagg (Cancer Research U.K. Centre for Cancer
(< 75 A2) trend, with an increase in the odds ratio to 10-fold using
Therapeutics) delivered a talk on the role of medicinal chemistry
as a reference point. Substitution to a free drug
design in the avoidance of toxicity at clinically effective exposures. In
analysis delivered a further rise to 27-fold.
setting the scene, clinical attrition drivers over a 10-year period span-
As a hypothesis, it was suggested that promiscuous binding to off-
ning 1991-2000 were highlighted (15). In contrast to a dramatic
target pharmacology was responsible for the increased incidence of
decline in attrition through pharmacokinetic aspects, the most
adverse outcomes associated with high CLogP/low TPSA space. To
prominent cause in 1991 to a minor bit player in 2000, drug failure
support the theory, CEREP data across the BioprintTM panel of 48
due to toxicology almost doubled to become a major factor entering
assays of varied target class (G protein-coupled receptors [GPCRs],
the 21st century. In addition a slight increase was also observed due
enzymes, ion channels) on 108 compounds were analyzed, using
to lack of clinical safety. However, Professor Blagg suggested pre-
> 50% inhibition at 10 µM at three or more targets as a definition of
clinical attrition rates due to safety and toxicity findings were likely to
promiscuity. In line with the observations derived at a toxicity level,
be significantly higher, given that such data often resided in confi-
compounds of CLogP > 3 and TPSA < 75 A2 appeared 25 times more
likely to have a significant off-target pharmacological profile than
Although mechanistically adverse outcomes could be linked to pri-
compounds of CLogP < 3 and TPSA > 75 A2. High lipophilicity has
mary pharmacology, secondary pharmacology or the presence of a
often been a design parameter used by the medicinal chemist in
structural alert or toxicophore, meta-analysis of in vivo tolerance
driving high potency against a target. Given its link to imparting an
(IVT) studies conducted on a dataset comprised of 245 potential
increased risk of toxicity and promiscuity, as well as high clearance,
Pfizer drug candidates accumulated over a 5-year period support a
Pfizer researchers have coined the term LipE, as defined by equation
further origin in the form of physicochemical drug properties (16).
(1), as a lipophilicity efficiency measurement that can be used by the
For each IVT study, the corresponding pharmacokinetic exposure
medicinal chemist to ensure increases in potency and compounddesign are directed away from liphophilicity risk factors associated
Cross-comparison of the chemical space property distribution map
(MW, CLogP, TPSA) of the IVT compound dataset with that of a
diverse subset of the Pfizer corporate file showed good overlap,
In relation to idiosyncratic adverse events triggered by chemical
thereby confirming that the set of compounds occupied sufficient
structure, in particular through metabolic activation, Professor
chemical diversity and the results of the meta-analysis hold general
Blagg focused on reducing dose size as a means of preventing attri-
application. Of the dataset, 50% were of a basic nature, 40% neutral
tion through this mechanism. The closely related analogues cloza-
pine (8) and olanzapine (9) served to illustrate the case (Fig. 2). Clozapine, at a clinical dose of 300 mg/day, is known to form reac-
Key to the meta-analysis was the importance of data deconvolution,
tive metabolites in vitro and cause a 1% incidence of argranulocyto-
and at what specific exposure threshold should intrinsic toxicity be
sis. In contrast to the restricted use of clozapine, olanzapine, dosed
was used as the single parameter to reflect exposure,
at 10 mg/day, shows no incidence of agranulocytosis, despite in vitro
and 10 µM total drug was selected as a pragmatic threshold level
studies confirming reactive metabolite formation.
that delivered a distribution of compounds between toxic and clean,while at the same time minimizing the number of compounds clas-
Furthermore, documented work has shown that drugs dosed at < 10
sified as uncertain. Data associated with compounds in which the
mg/day were associated with a significantly lower incidence of idio-
adverse in vivo outcome could correlate with the primary pharma-
syncratic drug reactions (18). Notwithstanding imparting the desired
THOMSON REUTERS – Drugs of the Future 2010, 35(1)
APPROACHES TO ASSESSING DRUG SAFETY IN THE DISCOVERY PHASE
also profiled during candidate selection for cardiovascular effects invivo using telemetered dogs. Patch clamp assays are clearly more
informative than binding assays, but as a manual assay throughputhas been limited. Newer, automated patch clamp robots (QPatch)
are now available and have markedly increased compound through-put per FTE. A further emerging technique with potential utility to be
deployed as part of a screening strategy is compound profiling in
spontaneously beating cardiomyocytes derived from human embry-
onic stem cells. Novartis investigates the effects of candidate com-pounds on cardiac action potentials in vivo using small animal mod-
els. Rabbits and guinea pigs respond similarly to humans in
response to sodium channel blockade, displaying prolonged P-R
and QRS intervals. During preclinical development, Novartis con-
ducts ECG profiling using telemetry in beagle dogs. Early sightingstudies are done using a noninvasive jacket telemetry system, with
studies in chronically implanted animals being undertaken prior to
Dr. Traebert briefly described a Novartis case study demonstratingthe utility of the aforementioned battery of assays to identify a com-
Figure 2. Structures of clozapine (8) and olanzapine (9).
pound with clear sodium channel liabilities that was dropped fromdevelopment. In vivo in dogs the compound affected P-R and QRSintervals, mean P wave duration, and also induced ventricular tachy-
intrinsic potency at the target, such a low projected clinical dose
cardia. Tachycardia was also observed in isolated rabbit heart. In
demands in conjunction superb pharmacokinetic and physicochem-
vitro the compound was active in cells stably transfected with the
ical properties, all of which remain under the remit and influence of
SCN5A gene. Dr. Traebert concluded by stressing the importance of
screening for sodium channel activity during compound develop-
The afternoon session of the symposium was opened by Dr. Martin
ment, adding that consideration of this area is underestimated com-
Traebert (Head of Safety Pharmacology EU, Novartis Institute for
pared to the investigation of compound activity against hERG chan-
Biomedical Research) with a presentation entitled “Addressing car-
nels. A variety of in vitro, ex vivo and in vivo assays are available, and
diac sodium channel liabilities during preclinical drug develop-
significant inhibition that translates into ECG alterations should be a
ment”. Dr. Traebert began by describing how, historically, pharma-
ceutical industry interest in cardiac sodium channels had centered
The second speaker in the afternoon session was Dr. James Dykens
on trying to develop inhibitors as antiarrhythmic agents. Encainide
(Director of Investigative Cellular Toxicity, Pfizer, U.K.). Dr. Dykens
and flecainide were two drugs progressed into clinical development
began his presentation by highlighting the problem caused by
and investigated in the Cardiac Arrythmia Suppression Trial (CAST).
adverse drug reactions, referencing data from the U.S. Over 2.2 mil-
Unfortunately, however, these drugs were shown to cause a 3.6-fold
lion adverse drug reactions occur annually in hospitalized patients
increase in arrhythmic death and a 2.5-fold increase in overall mor-
alone in the U.S., leading to approximately 106,000 deaths/year.
tality (19). Molecular biology studies have revealed that the human
Adverse drug reactions are the fourth leading cause of death in the
cardiac sodium channel contains four transmembrane repeats and
U.S. Dr. Dykens view was that serious drug toxicity liabilities were
is encoded by the gene SCN5A. The channel displays strictly volt-
clearly not being identified, and proposed that off-target drug
age-dependent activation, and is responsible for the depolarization
effects on mitochondria were important in this respect. Many drugs
phase and “upstroke” of the action potential. Inherited loss-of-func-
withdrawn from the market or receiving black box warnings have
tion mutations of SCN5A are associated with a range of chan-
been shown to impair mitochondrial function. The pharmaceutical
nelopathies, including congenital long Q-T syndrome, idiopathic
industry is now starting to research this area more closely using
ventricular fibrillation (Brugada syndrome), isolated cardiac conduc-
tion disease, atrial standstill, congenital sick sinus syndrome, sud-
Mitochondria are the “gatekeepers” of cell death. If mitochondria
den infant death syndrome and dilated cardiomyopathy.
die, then so does the cell. These organelles have evolved from
Currently, although there is now increased awareness of sodium
ancient bacteria, and mitochondrial DNA represents the only non-
channel liabilities by regulatory authorities, there are no formal
nuclear genome in all animals. Mitochondria generate > 90% of cel-
guidelines on this topic. Drug companies are, however, deploying a
lular energy, and the magnitude of this activity is highlighted by the
range of preclinical assays to identify compounds possessing sodi-
fact that human males turn over 193 lbs of ATP/day, while females
um channel liabilities. Novartis routinely uses a binding assay, with
turn over approximately 148 lbs/day. Studies have shown that drugs
rat brain as a sodium channel source, as a screen during lead selec-
can interfere with mitochondrial electron transfer and ATP genera-
tion. Moving towards candidate selection, compounds are tested in
tion at many points. A Pfizer study of 550 drugs revealed that 34%
repolarization assays using the patch clamp technique with HEK-
of drugs displaying organ toxicity impaired mitochondrial function.
293 or CHO cells stably transfected with SCN5A. Compounds are
Drugs impairing mitochondrial function will have an adverse effect,
THOMSON REUTERS – Drugs of the Future 2010, 35(1)
APPROACHES TO ASSESSING DRUG SAFETY IN THE DISCOVERY PHASE
although clinical manifestation is dependent on the bioenergetic
multiple measures of in vitro toxicity are used to aid compound opti-
capacity of an individual animal or patient. Bioenergetic capacity is
mization in order to reduce the attrition rate in the preclinical and
determined by genetics and also age. Indeed, older animals have
been shown to be more susceptible to troglitazone-induced toxicitythan younger healthy animals.
Dr. Dykens went on to discuss further his experience at Pfizer using
a range of assays to detect drugs possessing mitochondrial toxicity. The first assay described was a 96-well plate format mitochondrial
respiration assay (20, 21). This assay revealed potent inhibitory activ-
–Nongenotoxic carcinogenicity Human and rat AhR, rat PPARα
ity for a number of thiazolidinediones, and subsequent work with apioglitazone photoaffinity probe pulled down MitoNEET, an atypical
Glu, Calcein, Alamar Blue, Hoechst Luxcel
2Fe-2S protein, as a molecular target for this drug. A further mito-
chondrial functional assay utilizes “Seahorse technology”. This sys-
tem measures oxygen consumption rate and extracellular acidifica-tion rate in microchambers. Phenformin and butformin, drugs
1A1, 1A2, 2A6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4
withdrawn for causing lactic acidosis, were shown to be active in this
Throughput: 40-80 compounds per assay batch
assay, while metformin was inactive (22). Mitochondrial impairmentby thiazolidinediones and statins was also demonstrated using anassay format that measures the activity of individual oxidative phos-
For fast genotoxicity screening, a panel of six screens is used, all of
phorylation complexes following immunocapture (23).
which depend on sensitive luminescent luciferase-based assays. A
Contemporary cell culture conditions typically contain glucose con-
bacterial screen is performed in Salmonella, where the luciferase
centrations five times the physiological levels. Elevated glucose
expression is activated via a cascade of reactions known as the SOS
inhibits mitochondrial respiration (Crabtree effect), and consequent-
system, which is employed in the “Vitotox” assay to detect the geno-
ly, drugs possessing mitochondrial toxicity will not be detected
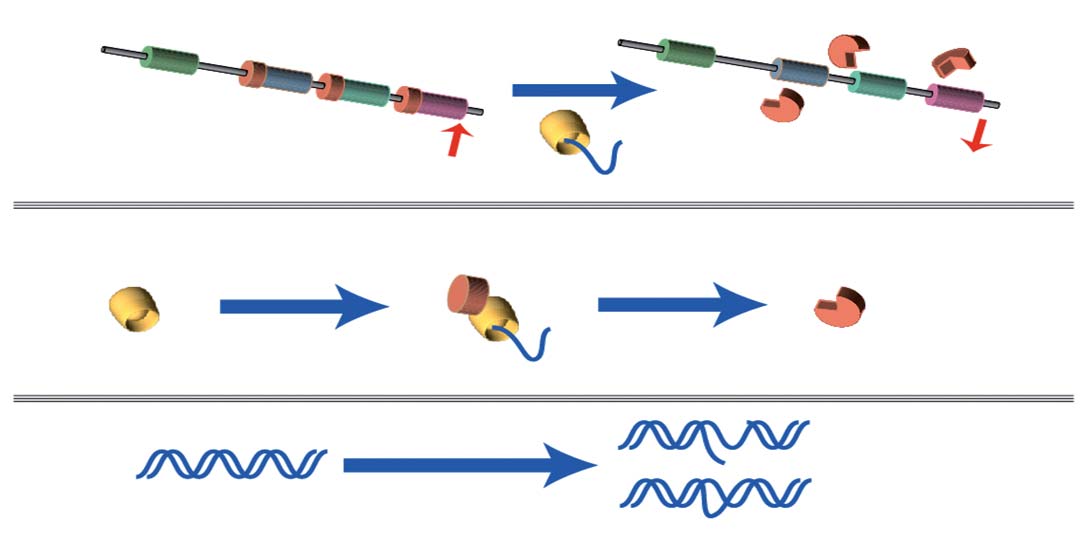
toxicity, cytotoxicity and mutagenic potency of the sample (Fig. 3).
under such conditions. However, cells grown in galactose do not
The major benefit of the assay is that the entire DNA content of the
generate ATP through glycolysis and become susceptible to drugs
cell functions as a target for the genotoxin to display its effect, and
inhibiting mitochondrial function (24). In terms of useful in vivo
can be considered as a substitute for the full Ames mutagencity test.
models, heterozygous superoxide dismutase 2 (SOD+/–) mice display
With respect to clastogenicity, a “RadarScreen” (reMYND, Leuven,
clinically silent mitochondrial dysfunction, and in contrast to normal
Belgium) assay is used to replace the sister chromatid exchange
mice are susceptible to the hepatotoxic effects of troglitazone (25).
(SCE), chromosomal aberration (CA) assays and micronuclei tests,
Dr. Dykens concluded his talk by reiterating his view that a key con-
which are time-consuming and have low compound throughput.
tributor to idiosyncratic drug responses is mitochondrial dysfunction.
The assay is based on activation of the RAD54 promoter linked to a
Individual susceptibility is determined by an individual’s “bioener-
β-galactosidase reporter gene in yeast, as shown below (Fig. 4).
getic threshold”. Pfizer has moved assessment of mitochondrial tox-icity to the lead selection phase of discovery, where there are poten-
Good sensitivity, selectivity and predictability for the clastogenicity
tially diverse hit series and the greatest opportunity to “de-risk” for
test are observed, although prediction for mutagenicity is relatively
hepatotoxicity, nephrotoxicity and neurotoxicity.
low, which is the opposite for the Vitotox assay. More mechanisticassays for genotoxicity in Hep G2 cells were described, in which four
The next speaker was Dr. Willem Schoonen (Department of Toxicol-ogy and Drug Disposition, Schering-Plough, the Netherlands), who
promoters of the luciferase assay were used. An advantage of these
presented his company’s approach to “High-Throughput Screening
cells is their ability to metabolize/activate certain drugs such as
for Toxicity Testing” in early drug discovery. Despite the efforts by the
benzo[a]pyrene, aflaxtoxin B1 and etoposide, without the need for
pharmaceutical industry over the last 15 years, some 50% of new
the S9 metabolic activating system (28-30). Although the cells do
chemical entities (NCEs) still fail due to toxicity, which represents a
not have CYP2C9 activity, it can be induced.
major cost to industry (15), particularly in the areas of hepatotoxici-
All of these assays were validated against a large number of geno-
ty, cardiotoxicity, skin toxicity, CNS side effects, genotoxicity and car-
toxic (and nongenotoxic) compounds (190) with diverse mecha-
cinogenicity. The preclinical cost is even more significant, as safety
nisms, including direct-acting genotoxins, topoisomerase inhibitors,
screening is often the final hurdle in drug discovery before the NCE
nucleotide/DNA synthesis inhibitors, reactive oxygen species gener-
enters the clinic, and few effective strategies for avoiding toxicity
ators and aneugenes (change in number of chromosomes). In gen-
exist to guide medicinal chemistry programs before then (26, 27).
eral, the sensitivity, specificity and predictivity of the assays are
Dr. Schoonen described the assays Schering-Plough uses to assess
acceptable, with a total of 110 compounds having a positive Ames or
DNA and membrane damage, cell-, organ- or organelle-specific tox-
clastogenicity score (31). It was concluded that Vitotox prediction is
icity, nuclear receptor activation and CYP enzymes and induction,
relatively high, RadarScreen is very good and prediction of all four
which are used to select/deselect and/or rank compounds in lead
different Hep G2 assays is relatively low in comparison with
optimization. What assay in which phase depends on sample
Salmonella and yeast genotoxicity assays. Nevertheless, in vitro
throughput, the amount of compound available and the effort
high-throughput screening might be more valuable for the predic-
required in conducting the assay. Multiple leads are screened and
tion of human genotoxicity (32, 33).
THOMSON REUTERS – Drugs of the Future 2010, 35(1)
APPROACHES TO ASSESSING DRUG SAFETY IN THE DISCOVERY PHASE
RecA cleavage of LexA repressor
Figure 3. Vitotox assay principle.
tool for the last 40 years, more recently they have been used as an in
vivo vertebrate model to support early drug discovery for screening
NCEs. The small size of the larvae makes them amenable to high-
throughput screening using very small quantities of compound.
Their metabolism, physiology and development are apparently com-
parable to humans, and since they are transparent, multiple end-
points can be visualized without dissection. Dr. Hill went on to
describe several assays that have been developed in conjunction
with a number of pharmaceutical companies, as detailed below
Uptake into zebrafish can vary considerably from compound to com-
pound, and no one physicochemical property can adequately predict
compound uptake through the skin. A generic protocol is therefore
employed to measure the uptake of compounds using conventional
Figure 4. RadarScreen assay principle.
bioanalytical methods, which helps to identify false negatives and to
correlate the extent of exposure with toxicity.
The last speaker of the day was Dr. Adrian Hill (Evotec, U.K.), who
Data were presented on a cardiac functional assay, where it was con-
gave a presentation on the use of zebrafish screening in early drug
cluded that the cardiophysiological response of zebrafish was
discovery, with reference in particular to predicting hepatotoxicity,
predictive of the human cardiovascular response. Some 64 com-
cardiotoxicity and developmental toxicity (embryotoxicity and ter-
pounds were used in the validation, a summary of which is present-
atogenicity). Although zebrafish have been used as an experimental
Table I. Multifunctional uses of zebrafish in drug discovery.
Developmental toxicity (embryotoxicity & teratogenicity)
THOMSON REUTERS – Drugs of the Future 2010, 35(1)
APPROACHES TO ASSESSING DRUG SAFETY IN THE DISCOVERY PHASE
Table II. Zebrafish endpoints associated with teratogenicicity.
–Atrioventricular decoupling: 31 of 36 (86%)
Two causing no cardiac effect (erythromycin, sotalol) – poor uptake
Three causing bradycardia (mibefradil, nortriptyline, ranolazine)
One causing atrioventricular decoupling (diltiazem)
–Overall predictivity = 91% (excluding bioanalysis data)
Information was also presented from a Novartis (blinded) study
using 20 compounds, in which bradycardia was correctly predictedfor 5 of 5 compounds. In terms of Q-T-prolonging drugs: 91% werecorrectly identified by atrioventricular decoupling (reducing to 71%due to false negatives with poor uptake). Excluding unclassified
DISCLOSURES
compounds, the overall predictivity amounted to 94%.
J. Allen, P. Jeffrey and A. Ratcliffe are in the paid employ of their
In terms of developmental toxicity (embryotoxicity and teratogenici-
respective companies. All authors are SMR Committee members for
ty), a number of institutions have used a zebrafish assay employing
different protocols and strains/ages of larvae. Endpoints associatedwith teratogenicity are detailed in Table II. Overall predictivity
REFERENCES
1. Rieder, M.J., Reiner, A.P., Gage, B.F. et al. Effect of VKORC1 haplotypes on
Finally, in terms of hepatotoxicity, a zebrafish assay was described
transcriptional regulation and warfarin dose. N Engl J Med 2005, 352(22):
using a number of endpoints, including liver abnormalities, changes
in size and shape of the liver (hepatomegaly), effects on yolk absorp-
2. Jorgensen, A.L., Al-Zubiedi, S., Zhang, J.E. et al. Genetic and environmen-
tion (yolk retention), lethality, gastrointestinal toxicity and loss of
tal factors determining clinical outcomes and cost of warfarin therapy: A
bile. The assay was compared to a novel cell-based model using a
prospective study. Pharmacogenet Genomics 2009, 19(10): 800-12.
high-content screening (HCS) model (34). In addition to 14 well-
3. Limdi, N.A., Veenstra, D.L. Warfarin pharmacogentics. Pharmacotherapy
known toxic compounds, a subset of 36 compounds from the HCS
assay was selected for screening including:
4. Norgard, N.B., Abu-Fadel, M. Comparison of prasugrel and clopidogrel inpatients with acute coronary syndrome undergoing percutaneous coronaryintervention. Vasc Health Risk Manag 2009, 5: 873-82.
5. Motovska, Z., Widimsky, P. Improving outcomes in patients undergoing
–Human hepatotoxic compounds that gave a false negative in the HCS
percutaneous coronary intervention: Role of prasugrel. Vasc Health Risk
(stavudine, novobiocin, bupropion, diethylcarbamazine, clofibrate,
6. Park, K.B., Dalton-Brown, E., Hirst, C., Williams, D.P. Selection of new
–Toxic compounds to animals that gave a false negative in the HCS (rido-
chemical entities with decreased potential for adverse drug reactions. Eur J
–Toxic compounds to humans but NOT liver toxic that gave a false posi-
7. Kocsis, Á.K., Szabolcs, A., Hofner, P., Takács, T., Farkas, G., Boda, K.,
tive in the HCS (metformin [pancreas], pamidronate [kidney], astemi-
Mándi, Y. Plasma concentrations of high-mobility group box protein 1, sol-
zole [acute], temozolomide [bone marrow], gentamicin)
uble receptor for advanced glycation end products and circulating DNA inpatients with acute pancreatitis. Pancreatology 2009, 9(4): 383-91.
–Nontoxic compounds: false positive in the HCS (picotamide)
8. Luft, T., Conzelmann, M., Benner, A. et al. Serum cytokeratin-18 fragmentsas quantitative markers of epithelial apoptosis in liver and intestinal graft-versus-host disease. Blood 2007, 110(13): 4535-42.
The HCS resulted in 6 of 9 false positives (gentamicin, metformin,
9. Park, K., Williams, D.P., Naisbitt, D.J., Kitteringham, N.R., Pirmohamed,
astemizole, temozolomide, picotamide, pamidronate) and 9 of 27
M. Investigation of toxic metabolites during drug development. Toxicol
false negatives (clofibrate, stavudine, novobiocin, bupropion,
Appl Pharmacol 2005, 207(2, Suppl.): 425-34.
diethylcarbamazine, pravastatin, ridogrel, acaftadine, valproate).
10. Merk, H.F. Diagnosis of drug hypersensitivity: Lymphocyte transformation
Overall, the HCS assay sensitivity was 67%, specificity 33% and pre-
test and cytokines. Toxicology 2005, 209(2): 217-20.
dictivity 58%. In comparison, the zebrafish assay gave 3 of 50 false
11. Mallal, S., Phillips, E., Carosi, G. et al. HLA-B*5701 screening for hypersen-
positives (gentamicin, praziquantel, astemizole) and 5 of 50 false
sitivity to abacavir. N Engl J Med 2008, 358(6): 568-79.
negatives (oxyphenisatin, valproate, ketoconazole, novobiocin,
12. Daly, A.K., Donaldson, P.T., Bhatnagar, P. et al. HLA-B*5701 genotype is a
stavudine), leading to an increased sensitivity of 86%, specificity of
major determinant of drug induced liver injury due to flucloxacillin. Nat
77% and overall predictivity of 84%.
THOMSON REUTERS – Drugs of the Future 2010, 35(1)
APPROACHES TO ASSESSING DRUG SAFETY IN THE DISCOVERY PHASE
13. Evans, D.C., Watt, A.P., Nicoll-Griffith, D.A., Baillie, T.A. Drug-protein
24. Marroquin, L.D., Hynes, J., Dykens, J.A., Jamieson, J.D., Will, Y. adducts: An industry perspective on minimizing the potential for drugCircumventing the Crabtree effect: Replacing media glucose with galactosebioactivation in drug discovery and development. Chem Res Toxicol 2004,
increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol
14. Nakayama, S., Atsumi, R., Takakusa, H. et al. A zone classification system
25. Ong, M.M.K., Calivarathan, L., Boelsterli, U.A. Troglitazone induced
for risk assessment of idiosyncratic drug toxicity using daily dose and cova-
hepatic necrosis in an animal model of silent genetic mitochondrial
lent binding. Drug Metab Dispos 2009, 37(9): 1970-7.
abnormalities. Toxicol Sci 2007, 97(1): 205-13.
15. Kola, I., Landis, J. Can the pharmaceutical industry reduce attrition rates?
26. Kramer, J.A., Sagartz, J.E., Morris, D.L. The application of discovery toxi-
Nat Rev Drug Discov 2004, 3(8): 711-6. cology and pathology towards the design of safer pharmaceutical leadcandidates. Nat Rev Drug Discov 2007, 6(8): 636-49.
16. Hughes, J.D., Blagg, J., Price, D.A. et al. Physiochemical drug propertiesassociated with in vivo toxicological outcomes. Bioorg Med Chem Lett
27. Stevens, J.L. Future of toxicology - Mechanisms of toxicity and drug safe-ty:where do we go from here. Chem Res Toxicol 2006, 19(11): 1393-401.
28. Westerink, W.M.A., Schoonen, W.G.E.J. Cytochrome P450 enzyme levels
17. Ryckmans, T., Edwards, M.P., Horne, V.A. et al. Rapid assessment of ain HepG2 cells and cryopreserved primary human hepatocytes and theirnovel series of selective CB2 agonists using parallel synthesis protocols: Ainduction in HepG2 cells. Toxicol In Vitro 2007, 21(8): 1581-91. lipophilic efficiency (LipE) analysis. Bioorg Med Chem Lett 2009, 19(15):
29. Westerink, W.M.A., Schoonen, W.G.E.J. Phase II enzyme levels in HepG2cells and cryopreserved primary human hepatocytes and their induction in
18. Uetrecht, J. Prediction of a new drug’s potential to cause idiosyncratic reac-HepG2 cells. Toxicol In Vitro 2007, 21(8): 1592-602. tions. Curr Opin Drug Discov Devel 2001, 4(1): 55-9.
30. Westerink, W.M.A., Stevenson, J.C.R., Schoonen, W.G.E.J. Pharmacologic
19. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. profiling of human and rat cytochrome P450 1A1 and 1A2 induction andPreliminary report: Effect of encainide and flecainide on mortality in a ran-competition. Arch Toxicol 2008, 82(12): 909-21.
dom trial of arrhythmia suppression after myocardial infarction. N Engl J
31. Westerink, W.M.A., Stevenson, J.C.R., Lauwers, A., Griffoen, G., Horbach,
G.J., Schoonen, W.G.E.J. Evaluation of the Vitotox and RadarScreen assays
20. Dykens, J.A., Marroquin, L.D., Will, Y. Strategies to reduce late stage attri-for the rapid assessment of genotoxicity in the early research phase of drugtion due to mitochondrial toxicity. Expert Rev Mol Diagn 2007, 7(2): 161-
development. Mutat Res 2009, 676(1-2): 113-30.
32. Knasmüller, S., Mersch-Sundermann, V., Kevekordes, S. et al. Use of
21. Will, Y., Hynes, J., Ogurtsov, V.I., Papkovsky, D.M. Analysis of mitochondr-human-derived liver cell lines for the detection of environmental and dietaryial function using phosphorescent oxygen sensitive probes. Nat Protoc
genotoxicants; current state of knowledge. Toxicology 2004, 198(1-3): 315-
22. Dykens, J.A., Jamieson, J., Marroquin, L., Nadanaciva, S., Billis, P.A., Will,
33. Mersch-Sundermann, V., Knasmüller, S., Wu, X.-J., Darroudi, F., Kassie, F.
Y. Biguanide-induced mitochondrial dysfunction yields increased lactateUse of a human-derived liver cell line for the detection of cytoprotectiveproduction and cytotoxicity of aerobically poised HepG2 cells and humanantigenotoxic and cogenotoxic agents. Toxicology 2004, 198(1-3): 329-
hepatocytes in vitro. Toxicol Appl Pharmacol 2008, 233(2): 203-10.
23. Nadanaciva, S., Dykens, J.A., Bernal, A., Capaldi, R.A., Will, Y.
34. O'Brien, P.J., Irwin, W., Diaz, D. et al. High concordance of drug-inducedMitochondrial impairment by PPAR agonists and statins identified viahuman hepatotoxicity with in vitro cytotoxicity measured in a novel cell-immunocaptured OXPHOS complex activities and respiration. Toxicol
based model using high content screening. Arch Toxicol 2006, 80(9):
Appl Pharmacol 2007, 223(3): 277-87.
THOMSON REUTERS – Drugs of the Future 2010, 35(1)
INSTITUTO MUNICIPAL DE PESQUISA, ADMINISTRAÇÃO E RECURSOS HUMANOS – IMPARH Prova aplicada em 18 de maio de 2008 (TARDE). PROVA DE ANESTESIOLOGISTA - SMS Este Caderno de Prova contém 40 (quarenta) questões, numeradas de 01 a 40, todas com 04 (quatro) alternativas. Verifique se o caderno está completo ou se há imperfeições. Nestes casos, 01. Sobre a hipotermia, é correto afirmar q
Washout Periods for Brimonidine for latanoprost ( n ؍ 17) was 4.4 ؎ 3.2 weeks ( P ؍ .24). 0.2% and Latanoprost 0.005% In all but one patient, brimonidine returned to baseline by 5 weeks and latanoprost returned by 8 weeks. William C. Stewart, MD, Keri T. Holmes, and CONCLUSION: After discontinuing latanoprost or bri- Mark A. Johnson monidine, a wide variation exist
 APPROACHES TO ASSESSING DRUG SAFETY IN THE DISCOVERY PHASE
RecA cleavage of LexA repressor
Figure 3. Vitotox assay principle.
APPROACHES TO ASSESSING DRUG SAFETY IN THE DISCOVERY PHASE
RecA cleavage of LexA repressor
Figure 3. Vitotox assay principle.