Do you want to buy antibiotics online without prescription? https://buyantibiotics24h.net/ - This is pharmacy online for you!
Pajoheshyar.pasteur.ac.ir
NIH Public Access Author Manuscript Gastroenterology. Author manuscript; available in PMC 2009 May 2. Gastroenterology. 2008 February ; 134(2): 405–415. doi:10.1053/j.gastro.2007.11.036. Virologic Monitoring of Hepatitis B Virus Therapy in Clinical Trials and Practice: Recommendations for a Standardized Approach JEAN–MICHEL PAWLOTSKY*,‡, GEOFFREY DUSHEIKO§, ANGELOS HATZAKIS||, DARYL LAU¶, GEORGE LAU#, T. JAKE LIANG**, STEPHEN LOCARNINI‡‡, PAUL MARTIN§§, DOUGLAS D. RICHMAN|| ||, and FABIEN ZOULIM¶¶
* French National Reference Center for Viral Hepatitis B, C and delta, Department of Virology, Henri MondorHospital, University of Paris XII, Créteil, France ‡ INSERM U841, Créteil, France § Centre for Hepatology,Royal Free and University College School of Medicine, London, United Kingdom || Department of Hygiene,Epidemiology and Medical Statistics, Athens University Medical School, Athens, Greece ¶Liver Center,Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston,Massachusetts # Department of Medicine, Queen Mary Hospital, University of Hong Kong, Hong Kong SAR,China **Liver Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, NationalInstitutes of Health, Bethesda, Maryland ‡‡ Victorian Infectious Diseases Reference Laboratory, NorthMelbourne, Victoria, Australia §§ Division of Liver Diseases and Recanati-Miller Transplant Institute, MountSinai School of Medicine, New York, New York || || VA San Diego Healthcare System and University ofCalifornia San Diego, La Jolla, California ¶¶ INSERM U871, Hospices Civils de Lyon, Department of LiverDiseases, Université Lyon 1, Lyon, FranceAbstract
Treatment of chronic hepatitis B virus (HBV) infection is aimed at suppressing viral replication tothe lowest possible level, and thereby to halt the progression of liver disease and prevent the onsetof complications. Two categories of drugs are used in HBV therapy: the interferons, includingstandard interferon alfa or pegylated interferon alfa, and specific nucleoside or nucleotide HBVinhibitors that target the reverse-transcriptase function of HBV-DNA polymerase. The reportedresults of clinical trials have used varying definitions of efficacy, failure, and resistance based ondifferent measures of virologic responses. This article discusses HBV virologic markers and tests,and their optimal use both for planning and reporting clinical trials and in clinical practice.
Hepatitis B virus (HBV) infection is a major public health problem, with approximately 350million individuals chronically infected worldwide.1 Individuals with chronic hepatitis B areexposed to a risk of complications such as cirrhosis, hepatic decompensation, andhepatocellular carcinoma.2 Treatment of chronic hepatitis B is aimed at suppressing viralreplication to the lowest possible level, and thereby to halt the progression of liver disease andprevent the onset of complications. However, HBV infection cannot be eradicated fully becauseof the persistence of covalently closed circular DNA (cccDNA) in the nuclei of infectedhepatocytes. Two categories of drugs are used in HBV therapy: (1) standard or pegylatedinterferon (IFN) alfa, and (2) specific nucleoside or nucleotide HBV inhibitors that target thereverse-transcriptase function of HBV-DNA polymerase. Four HBV inhibitors currently areapproved for HBV therapy in the United States, Europe, and most Asian and Latin Americancountries: lamivudine, adefovir dipivoxil, entecavir, and telbivudine. Tenofovir disoproxil
Address requests for reprints to: Jean-Michel Pawlotsky, MD, PhD, Department of Virology, Hopital Henri Mondor, 51 Avenue duMaréchal de Lattre de Tassigny, 94010 Crtéil, France. e-mail: E-mail: jean-michel.pawlotsky@hmn.aphp.fr; fax: (33) 1-4981-4831.
fumarate and the combination of tenofovir and emtricitabine have potent activity against HBV,but to date are approved only for use in the treatment of human immunodeficiency virus
infection. Clinical trials are ongoing to assess their utility and the utility of other new anti-HBVdrugs for the treatment of HBV infection.
The goal of treatment with specific HBV inhibitors is to produce an antiviral effect that is asprofound and as sustained as possible to efficiently prevent the complications of chronic HBVinfection in the long term. Chronic administration of HBV inhibitors frequently leads to viralresistance, particularly with incomplete suppression of HBV replication. The selection of HBVvariants with amino acid substitutions in the reverse-transcriptase domain of HBV-DNApolymerase confers reduced susceptibility to the inhibitory action of the drug.3,4 Resistanceis a major issue in clinical practice because it leads to HBV treatment failures and progressionof liver disease.5
Context and Objectives
Clinical trials of therapies of HBV infection have led to current drug approvals, including IFNsand nucleos(t)ide analogues. The reported results have used varying definitions of efficacy andfailure based on different measures of virologic responses. In patients with hepatitis B e antigen(HBeAg)-positive chronic hepatitis B, trials generally report rates of HBeAg loss and HBeAgseroconversion, alanine aminotransferase (ALT) normalization, and suppression of serum
HBV DNA. Trials of conventional IFN alfa6,7 and the early lamivudine studies8 –10 reportedsuppression of serum HBV DNA as measured by hybridization-based methods, which haddetection limits of around 105 copies/mL. With the advent of more sensitive assays forquantification of serum HBV-DNA level, recent trials with both pegylated IFNs and nucleos(t)ide analogues have used a variety of definitions of serum HBV-DNA response with levelsof suppression ranging from less than 500,000 copies/mL to less than 300 copies/mL.11–19In patients with HBeAg-negative chronic hepatitis B, HBeAg seroconversion is not the endpoint: a combined end point of biochemical response (ALT normalization) and virologic(serum HBV-DNA suppression) response is used frequently. However, inconsistent levels ofthe target serum HBV-DNA level have been chosen. In addition, different results are reportedin different units depending on the assay used. In clinical practice, different assays may beused, even for sequential assays for the same patient, making interpretation of results andidentification of the emergence of resistance difficult.
Definitions and hence the reporting of “resistance” across clinical trials also vary. In somecases, the incidence of genotypic mutations (ie, nucleotide alterations that result in amino acidsubstitutions that are selected by antiviral drugs) may be reported with no reference to whether
the mutations correlate with any virologic rebound (increase of levels of serum virus in aresponder patient) or effect on clinical or biochemical parameters. Conversely, virologic andbiochemical breakthrough may be reported with no description of associated viral mutationand pharmacologic data.
As more antiviral therapies become available for the treatment of chronic hepatitis B, the riskof emergence of resistance and cross-resistance will increase, and as more options for managingpatients with antiviral drug resistance are developed, it will become important to define,understand, and be able to use and interpret the results of HBV virologic tools in themanagement of HBV therapy. The aim of this article is to discuss the virologic markers andtests and their optimal use both when planning and reporting clinical trials and in clinicalpractice. The authors met for 2 days. Four questions were discussed: what HBV markers shouldbe used and what is their utility in clinical trials and practice? What are the definitions oftreatment responses and failures and how should they be assessed virologically? How shouldHBV treatment be managed with virologic tools in clinical trials? How should HBV treatment
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
be managed with virologic tools in clinical practice? A consensus was reached on each pointafter extensive discussion. A draft summary of the group’s conclusions was circulated and
finalized with every author’s comments and suggestions. In this article, evidence-based (EB)recommendations are identified and recommendations based on the experts’ opinions (EO) arepresented. The authors acknowledge that virologic testing is expensive and not readilyavailable or affordable in many countries where hepatitis B is prevalent. Therefore, therecommendations for clinical practice should be considered best practice. HBV Markers and Their Utility in HBV Clinical Trials and Practice
The accepted virologic and biochemical markers (HBeAg, anti-HBe antibodies, serum HBVDNA, and serum ALT) used in the diagnosis and monitoring of HBV disease clearly are usefulfor the evaluation of patients both in clinical trials and in clinical practice (EB). Otherpotentially useful markers include liver cccDNA and quantitative hepatitis B surface antigen(HBsAg), HBV genotype, and genotypic resistance markers (Table 1). HBeAg/Anti-HBe Antibodies, Serum HBV DNA, and ALT
Among HBeAg/anti-HBe antibodies, serum HBV DNA, and ALT, the best viral marker forthe management of HBV disease (including both HBeAg-positive and HBeAg-negative) isserum HBV DNA. Serum HBV-DNA level is an indicator of disease prognosis. Several studies
have shown that increasing HBV viral level, starting at 104 copies/mL, is a predictor of riskfor the development of cirrhosis and hepatocellular carcinoma, regardless of HBV genotype,HBeAg serostatus, and baseline serum ALT level.20 –22 It is unclear, however, if these datafrom a large Taiwanese cohort study of mostly HBeAg-negative patients can be generalizedto other populations of patients chronically infected with HBV, such as those with adult-acquired infection, or to the individual patient. In addition, serum HBV DNA cannot be usedfor prognostication in individual patients and the clinical context is important for the decisionregarding when to start treatment.
Importantly, the control of HBV replication and treatment outcome are correlated. Reductionof serum HBV-DNA level is associated with an increased rate of HBeAg seroconversion inHBeAg-positive patients.5,6,9,14 –19,23–25 However, with nucleoside or nucleotideanalogues, more potent HBV-DNA suppression is associated with only a small increase inHBeAg seroconversion. Reduction of HBV-DNA levels also is associated with higher rates ofhistologic response and lower rates of complications of liver disease.5,6,9,14–19,23–25 Forinstance, continuous reductions in the levels of HBV DNA with lamivudine delays clinicalprogression in patients with advanced fibrosis or cirrhosis and significantly reduces theincidence of hepatic decompensation.5 Measurement of the HBV-DNA level is critical for the
early detection of treatment failure that may be related to poor adherence to therapy or selectionof a resistant virus.5,18,26,27 In addition, viral kinetics while on therapy predict the emergenceof resistance. The likelihood of resistance to nucleos(t)ide analogues is very low when HBV-DNA level is undetectable (<300 copies/mL) during therapy. It is significantly higher inpatients with more than 103 copies/mL at week 24 or 48 of therapy and increases proportionallyto the HBV-DNA level (note that the nonstandardized copy/mL unit has been used in thepublished trials).18,27–29
Liver HBV cccDNA and Quantitative HBsAg
The persistence of HBV in the liver, despite antiviral therapy, is owing to the maintenance ofHBV cccDNA in the nuclei of infected cells. cccDNA levels have been assessed in liver biopsyspecimens from patients in the different phases of chronic HBV infection, both during and afterantiviral therapy, using selective polymerase chain reaction (PCR) assays for HBV cccDNAin liver biopsy specimens.30 –34 HBeAg-positive patients have a higher cccDNA copy number
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
per cell than do HBeAg-negative patients, and cccDNA levels correlate with the phase of HBVinfection. Patients who achieve HBeAg seroconversion during antiviral therapy have lower
baseline levels of cccDNA than do nonseroconverters, and cccDNA is reduced significantlyin patients who received long-term nucleos(t)ide therapy compared with placebo.30 –36 Inaddition, a significant reduction in serum HBsAg titer has been observed with adefovirdipivoxil, which correlated with changes in cccDNA level, total intracellular HBV-DNA level,and serum HBV-DNA level.30 Because cccDNA is the major template for transcription andtranslation of viral antigens, including HBsAg, this result suggests that change in serum HBsAgtiter might be used as a surrogate for change in liver cccDNA, the latter requiring a liver biopsy.
37 However, this suggestion requires validation. Standardization of these assays is now needed.
Levels of hepatic cccDNA may provide a greater predictive value of response than alternativemeasures for a range of clinical therapies. In a trial of IFN–lamivudine combination therapyvs lamivudine monotherapy, liver HBV cccDNA levels at the end of therapy gave a higherpredictive value for sustained virologic response than did serum HBV-DNA level or totalintrahepatic HBV-DNA level.31 Similarly, in patients with positive HBsAg and lymphoma,cccDNA levels before chemotherapy predicted reactivation of HBV after chemotherapy, withan optimal cut-off level of approximately 3 copies/cell predicting no reactivation.38 Larger-scale trials are required to determine if quantification of cccDNA may provide an indicator ofthe efficacy of antiviral therapy and an independent predictor of outcome. For the immediate
future, it is recommended that cccDNA assays and quantitative HBsAg assays be included asresearch tools to increase our understanding of clinical trial results (EO) (Table 1). HBV Genotypes
HBV is classified into 8 HBV genotypes, A–H, based on an 8% or more DNA sequencedifference over the whole genome.39,40 Data suggest that HBV genotype may be related todisease outcome. In Asia, genotype C is associated with a higher risk of reactivation of hepatitisB and progression to cirrhosis than genotype B.41,42 In Europe, genotype D is associated withmore active disease than the other genotypes, but the fact that genotype D also is associatedwith long-standing infection (as a result of acquisition at a younger age) may constitute a bias.
39,43,44 Genotype F was implicated recently in an enhanced risk of hepatocellular carcinomain Alaskan natives.45 Genotype also may influence response to IFN-based therapy. Afterpegylated IFN therapy, the rate of HBeAg loss at the end of the follow-up period for genotypesA, B, C, and D, respectively, was 47%, 44%, 28%, and 25%.46 A similar relationship wasobserved with the rate of HBsAg loss in the same study.47 In another study, the rate of HBeAgloss at the end of the follow-up period for genotypes A and D was 52% and 22%, respectively. However, the role of HBV genotype in predicting clinical outcomes, including therapy, remainsto be established. It is recommended that all clinical trials collect HBV genotype data, and
consideration should be given to stratifying trials according to genotype (EO) (Table 1). Inclinical practice, the positive and negative predictive values of the HBV genotype on diseaseprogression and treatment outcome have not been determined at the individual patient level,but genotype determination may become more important in the future as the data matures. Genotypic Resistance Markers
Sensitive assays are available that can detect resistant viral variants during therapy before anincrease in HBV-DNA level.48 –56 Early detection of genotypic change allows one to switchto alternative therapies and to avoid virologic rebound and hepatitis flare. This is particularlyuseful in patients with cirrhosis. The value of early detection of resistance by genotyping isless clear for patients without significant hepatic fibrosis, in whom serum HBV-DNAmonitoring may be adequate to diagnose the development of antiviral drug resistance. In thesetting of virologic breakthroughs, detection of resistance by genotyping can be used todistinguish between medication noncompliance and the selection of resistant variants. Gastroenterology. Author manuscript; available in PMC 2009 May 2.
Systematic testing for resistance by genotyping is mandatory in clinical trials to understandfully the properties of new therapeutic agents (EB). Recommendations for the frequency of
resistance testing in both clinical trials and clinical practice are discussed later. Virologic Assessment of Treatment Responses and Resistance Standardization of Quantification Units
Serum HBV-DNA levels are reported in many different units depending on the method usedand the manufacturer of the assay (eg, copies/mL, genome equivalents [Eq]/mL, mega-equivalents [MEq]/mL, or international units [IU]/mL). The World Health Organization hasdefined an international standard for HBV DNA nucleic acid amplification techniques57 thathas been used to calibrate the IU/mL. Several HBV-DNA quantification assays are availablethat have been normalized to the World Health Organization international standard.58 SerumHBV-DNA levels now should be expressed universally in IU/mL in all available assays toensure comparability between the assays, between different trials in which different assayshave been used, and to allow the creation of guidelines that can be applied to whatever assaywas used (in general, an IU is equivalent to approximately 5– 6 copies, depending on the assay)(EB). HBV-DNA Quantification Technology
With several HBV-DNA quantification assays available and with fund providers/insurerssometimes dictating which assay is to be used by a laboratory, it is important to recommendthe required properties of the test rather than any specific technology (Table 2). An assay witha lower limit of detection of 103 IU/mL may be sufficient to monitor and manage the patientbut, in some instances, for example, in patients with a low baseline HBV-DNA level or withprofound inhibition or viral replication during therapy, a more sensitive assay with a lowerlimit of detection of the order of 10 IU/mL may be required, to ensure detection of theemergence of resistance as early as possible (EB). A dynamic range of quantification of at least5 log10 is recommended and samples with an HBV-DNA level above the upper limit ofdetection of the assay should be diluted and retested to provide an end point. If no dilution isperformed, the result should be reported as higher than the upper limit of detection, but thiswill not allow monitoring of primary response to therapy and primary treatment failure. Forall of these reasons, real-time PCR quantification assays now strongly are recommended overother technologies, especially in clinical trials, because they are very sensitive and have a broaddynamic range of quantification (7– 8 log10 with the current assays) (EB).59 – 65 In addition,the assay used should be proven to quantify equally and accurately all HBV genotypes. It isimportant to use the same assay for a given patient in clinical practice. In the case of an assayswitch during a clinical trial or a cohort study of treated patients, the initial samples should be
retested with the new assay. This also ideally should be performed in clinical practice if theinitial sample(s) has (have) been stored. If not, HBV-DNA changes should be interpretedcautiously. HBV Genotyping Technology
The reference method for HBV genotype determination is sequencing followed byphylogenetic analysis of generated sequences together with reference sequences.59 This is theonly method suitable for the analysis of new genotypes or recombination between genotypesbut it is time consuming. Reverse hybridization techniques have proven very useful in clinicaltrials. They also can identify mixed genotype infections.59 Real-time PCR or multiplex PCRare potential alternatives if appropriately validated against the gold standard (ie, sequencing). Gastroenterology. Author manuscript; available in PMC 2009 May 2. HBV Resistance Testing by Genotyping
Direct sequencing-based techniques are the gold standard because all mutations can be
detected, which is particularly important with the increasing number of mutations reported. Incontrast, hybridization assays can detect only known specific mutations and require new probesto detect novel mutations.49 –56,59 In clinical trials, direct sequence analysis must be used. Testing for HBV mutations also should evaluate the proportion of the mixed mutant and wild-type populations, using at least the data from direct sequencing (also called populationsequencing), but more sensitive and quantitative results can be obtained from the sequencingof multiple clones.54,56,67– 69 Hybridization-based methods have the advantage of detectingresistant variants when they are present as minor populations (down to 10% of the total viralpopulation).55 More sensitive technologies, such as those based on mass spectrometry, arecurrently in development.50 They will be able to detect smaller proportions of viral variantsin complex viral mixtures, but the utility of such sensitivity remains to be determined. Real-time PCR-based techniques are potential alternatives,66 but they may not be suited yet toclinical trials and clinical practice given the high number of substitutions of interest andvariability among wild-type sequences. Phenotypic Resistance Testing
Phenotypic analysis can determine in vitro inhibitory concentrations (IC) of specific HBV
inhibitors (ie, it allows the testing of the susceptibility of a given HBV polymerase sequenceto the antiviral action of a given drug). Methods based on transient transfection and continuousprotein expression as well as transduction with recombinant HBV baculoviruses have beendescribed.70 –75 Testing by phenotype permits the quantification of the magnitude ofresistance to a drug and can interrogate for resistance without the need to know the responsiblemutations. Phenotypic analyses can confirm and assess the drug susceptibility associated witha given amino acid substitution and cross-resistance to other drugs from the same or otherfamilies. However, one must be careful in interpreting the results of in vitro phenotypic analysesbecause the replication properties observed in vitro may not always translate in vivo where thevirus is under the influence of a much more complex replicative environment.54,56,68,69
The IC50 is defined as the drug concentration that reduces replication in the in vitro model by50%. A fold change in IC50 can be considered significant if it is greater than the naturalvariability of the in vitro assay. However, the IC50 has been shown to vary from 2- to 5-foldup to more than 50- to 100-fold with amino acid substitutions known to be associated with invivo resistance to different drugs relative to the wild-type sequence.70,76 Indeed, the in vivopharmacodynamics of the drugs may have a significant impact on whether small variations inIC50 may translate into clinical resistance. It is not possible to predict with confidence what
the impact of reduced susceptibility in vitro will have on the response to a given drug. Asignificant fold change in IC50 defines resistance clinically if it is associated with a diminishedtreatment efficacy in vivo. Thus, clinical trial data are necessary to determine a level ofresistance that will impact response and thus guide treatment decisions. Phenotypic assays alsoare critical to provide information on cross-resistance. Virologic Definitions Definition of Baseline Viral Level
A baseline viral level is needed as a reference against which to assess treatment response ornonresponse. The baseline viral level ideally should be defined as the viral level taken within24 hours before the patient starts treatment (EO). This is distinct from any previous viral levelmeasurements performed to inform the decision to commence treatment. In the case of HBV-DNA fluctuations (more frequent in HBeAg-negative patients), this baseline HBV-DNA levelis the viral level against which therapy will be assessed in the subsequent weeks and months. Gastroenterology. Author manuscript; available in PMC 2009 May 2.
In clinical practice, it may not be practical to obtain the baseline viral level and the viral leveldetermination closest to the start of therapy should be used (EO). Definition of Treatment Antiviral Effect and Efficacy and of End Points Achieved by Therapy
Three levels of virologic response can be defined: antiviral effect, antiviral efficacy, and endpoint. Antiviral effect—When antiviral treatment is started, it is important to have an early indication that the patient is responding to therapy. An antiviral effect is defined as a 1 log10 IU/mL or greater reduction of serum HBV-DNA level from baseline within 3 months of starting therapy. Antiviral efficacy—The quantitative log10 reduction relative to baseline is a measure of antiviral treatment efficacy and the aim is to reduce HBV DNA to as low a level as possible to avoid resistance, to increase the possibility of HBeAg loss within the first or second year of treatment in HBeAg-positive patients, and to ensure adequate virologic suppression that then will lead to histologic improvement in all patients. The ability to quantify treatment efficacy by measuring the log10 reduction relative to baseline is dependent on the baseline viral level and the lower limit of detection of the HBV-DNA assay used. Treatment efficacy also can be defined as the ability of a given therapy to achieve an undetectable HBV-DNA level in a given
HBV-DNA assay. In clinical trials, treatment efficacy should be assessed by measuring boththe mean or median log10 reduction of HBV-DNA level and the proportion of patients withundetectable HBV DNA (less than the threshold of the assay, see previously for technicalrequirements) at various time points in all treatment groups (EO). End point—The goal of HBV therapies is to stop or slow the progression of liver disease to prevent cirrhosis, decompensation of cirrhosis, or hepatocellular carcinoma. Current therapies fall into 2 categories: IFN and pegylated IFN, and specific HBV nucleoside or nucleotide inhibitors. Patients also can be divided into whether or not they have a sustained response off treatment. The sustained response off treatment is defined as sustained HBe seroconversion, HBV-DNA level reduction, and ALT normalization in HBeAg-positive patients (EB). There is a recent controversy as to whether HBeAg seroconversion is an adequate end point for patients infected during infancy or childhood.77,78 These patients probably need permanent suppression of HBV DNA to levels undetectable by sensitive PCR assays and reduction of ALT levels to less than 0.5 times the upper limit of normal.77 In patients with HBeAg-negative chronic hepatitis B, the sustained response off treatment is defined as sustained HBV-DNA level reduction and ALT normalization. However, current evidence suggests that sustained virologic responses may be rare in HBeAg-negative patients (EB). This term may be more
applicable to IFN treatment because of the finite duration of therapy. For nucleos-(t)ideanalogues, treatment duration tends to be extended and the goal of therapy is to achieveprofound and sustained inhibition of HBV replication in both HBeAg-positive and HBeAg-negative patients. Studies have suggested that nucleoside analogue therapy can be stopped 6–12 months after HBe seroconversion in HBeAg-positive patients (EB). However, theguidelines for HBeAg-negative patients have not been defined. The ultimate goal of therapyfor all patients is HBsAg seroconversion (EO).
Standardization of the end points achieved by therapy is needed in both clinical trials and inclinical practice. Three different end points can be defined to classify patient outcome. Thesedo not signify that therapy can be stopped but serve to define end points as a goal that has beenachieved by therapy, at any time. First, in HBeAg-positive patients, the goal is to achieveHBeAg seroconversion, ideally with short-term therapy. In addition, in both HBeAg-positiveand HBeAg-negative patients the aim is to have sustained inhibition of viral replication to
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
improve liver disease and to avoid the development of resistance. The end points are as follows:(1) in HBeAg-positive patients: HBeAg seroconversion (HBeAg loss and gain of anti-HBe),
sustained inhibition of viral replication (serum HBV-DNA level <2000 IU/mL or any lowerthreshold), and normalization of serum ALT level; (2) in HBeAg-positive patients with noHBeAg seroconversion: sustained inhibition of viral replication (serum HBV-DNA level<2000 IU/mL or lower) and normalization of serum ALT level; (3) in HBeAg-negativepatients: sustained inhibition of viral replication (serum HBV DNA at most <200 IU/mL) andnormalization of serum ALT level. In all instances, the HBV-DNA thresholds represent aminimal efficacy end point. With the more sensitive real-time PCR-based HBV-DNA assaysand more potent anti-HBV drugs, HBV-DNA levels should be as low as possible and, ideally,undetectable (ie, below the lower limit of detection of the assays, which is of the order of 10IU/mL) to ensure full prevention of liver disease progression and complications and theemergence of resistance.77
Definition of Treatment Failure
Because of the different mechanisms of action of nucleos(t)ide analogue-based and IFN-basedtherapies, separate definitions of treatment failure are needed (EO). Furthermore, a definitionof treatment failure with nucleos(t)ide analogue-based therapy aims to identify thedevelopment of resistance, a problem that does not occur with IFN.
Nonresponse to antiviral treatment with nucleos(t)ide analogues or primary antiviral treatmentfailure is the failure to achieve more than 1 log10 decrease from base-line within 3 months ofstarting therapy. In some patients, a suboptimal response to therapy may be observed,characterized by a more than 1 log10 but less than 2–3 log10 IU/mL decrease at month 3 oftherapy.19 Secondary antiviral treatment failure is defined by a rebound of serum HBV-DNAlevels of 1 log10 IU/mL or greater from nadir in patients with an initial antiviral treatment effectas confirmed by 2 consecutive determinations at a 1-month interval.79 The main causes ofprimary and secondary antiviral treatment failure are poor adherence to therapy, lack of thedrug’s antiviral effect related to metabolic causes (problems with absorption, bioavailability,metabolism of prodrug to active metabolite, or phosphorylation of the antiviral agent to itstriphosphate), and selection of drug-resistant HBV mutants.80 – 82 Further research inpharmacogenetics and inherent metabolic alterations in bioavailability or differences in therate of phosphorylation of nucleosides, similar to studies performed with antiretroviral oranticancer drugs,83,84 may be important for understanding differences in primary response.
Although IFN therapy is directed toward HBeAg seroconversion, only about a third of selectedpatients achieve HBeAg seroconversion (ie, the majority do not seroconvert).12,23 Treatmentwith IFN is normally for 6 –12 months and seroconversion may occur after treatment cessation.
Hence, for IFN-based therapy, treatment failure is defined as failure to achieve a 1 log10 orgreater reduction from baseline within 6 months of starting therapy or HBe seroconversionduring treatment and follow-up evaluation (EO). This is based on experience with conventionalIFN, is preliminary, and will require more data from studies with the pegylated IFNs.12
The cumulative incidence of primary and secondary treatment failures should be reportedsystematically in clinical trials according to the formula shown later (EB). Definition of Resistance
Antiviral drug resistance reflects the reduced susceptibility of a virus to the inhibitory effectof a drug85 and results from a process of adaptive mutations in the HBV polymerase gene. Inclinical trials and practice, resistance is defined as the selection of variants bearing amino acidsubstitutions conferring reduced susceptibility to drug that result in primary or secondarytreatment failure (see earlier definition). Although it is more likely that resistance is identified
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
owing to secondary treatment failure, resistance may become a cause of primary treatmentfailure either because of transmission of resistant HBV or because of cross-resistance resulting
from previous therapies. In clinical practice, clinicians likely will be monitoring HBV-DNAlevels and failure to respond to treatment will be the first indication of the emergence ofresistance.
In clinical trials, the reporting of resistance has varied tremendously from one trial to another,thus considerably biasing the interpretation of the results and our knowledge of resistanceincidence with a given anti-HBV drug.5,10,14,18,26,27,86 –90 The incidence of resistance,which now is assessed by sequencing, should be reported as a cumulative probability ofoccurrence.18 In all clinical trials, the cumulative probability of the onset of (1) resistance, (2)resistance with virologic (HBV DNA) breakthrough, and (3) resistance with virologic (HBVDNA) and biochemical (ALT) breakthroughs, should be reported every year according to theduration of follow-up evaluation (EB). Calculation should be made by means of the followingformula:
where P is the cumulative probability that the event will occur, nx is the number of cases atyear x, and Nx is the number of patients still followed up at year x. For example, this formula
has been used to calculate the cumulative incidence of mutation selection over 5 years ofadefovir administration in patients with HBeAg-negative chronic hepatitis B.18 Clinical trialsin which subsets of patients systematically stop treatment might create bias in results dependingon what category of patient (responders or nonresponders) is taken off the trial. This thereforeshould be avoided in the design of clinical trials, especially with new drugs.
In addition to reporting resistance according to the earlier definition, it also would be useful toreport the number of patients with virologic and biochemical breakthrough without a viralcause (EO). If a mutation is identified in a patient, follow-up evaluation is important becausethe virologic breakthrough can be delayed several weeks after the appearance of the viralmutant. In trials of new drugs it is important to evaluate the emergence and incidence of newmutations and then the time to loss of response, defined as an increase (1 log10) in HBV-DNAlevel. However, some mutations may show phenotypic resistance but not confer clinical failurewithin the time-frame of a clinical trial. Management in Clinical Trials and Clinical Practice How Do We Monitor Treatment Efficacy and Failure in Clinical Trials and Clinical Practice?
To aid standardization of reports and comparison of different clinical trials with IFN and/ornucleos(t)ide HBV inhibitors, assessments of HBV DNA, ALT, and HBeAg/anti-HBe shouldbe performed at baseline and then every 1–3 months; although for a new drug, serum HBVDNA and ALT should be measured monthly to evaluate viral kinetics and incidence of ALTincreases (EO) (Table 3). In clinical practice, 3- to 6-month assessments are adequate (EB). After the baseline assessment, the first assessment at month 3 will allow evaluation of theprimary treatment response. In both clinical trials and clinical practice, if a serum HBV-DNAmeasurement indicates that the patient may have primary or secondary treatment failure, butthere is no increase in serum ALT level, a second serum HBV-DNA sample should be assayedfor confirmation 1 month later (EB). How Do We Show Resistance in Clinical Trials and Clinical Practice?
In clinical trials with nucleos(t)ide HBV inhibitors, direct sequencing of the polymerase geneshould be performed systematically at baseline, at the end of each year, at 3 months if there is
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
primary treatment failure, and at the time when secondary treatment failure is documented(EO) (Table 4).
In clinical practice, nucleos(t)ide analogue resistance testing based on direct sequencing orreverse hybridization on a baseline sample (if available) is required only in patients who havefailed on their current treatment (EB). If primary or secondary treatment failure has been shown,non-HBV–related causes of failure such as noncompliance should be eliminated, for instance,by measuring circulating drug concentrations. Resistance testing should be performed to showresistance and to inform the treatment decision (EO). Similar to the clinical trial setting, itshould be performed at 3 months in the case of primary treatment failure and at the time ofsecondary treatment failure (Table 4). However, consensus guidelines should be issued to guidetreatment decisions according to the patient’s genotypic resistance profile. How Do We Explore the Mechanisms of HBV Resistance in Clinical Trials?
Phenotypic assays and viral quasispecies analysis are useful in clinical trials with nucleos(t)ide HBV inhibitors and as a research tool (EO). Phenotypic assays are used to establish theconcentration of the drug that inhibits in vitro replication of HBV variants by 50% and 90%(IC50 and IC90). These numbers quantify the level of resistance to a specific drug conferred byviral mutation(s). Phenotypic assays can use nearly full-length HBV polymerase sequencesretrieved from patients before or during therapy, or prototype polymerase sequences into which
resistance substitutions have been incorporated by mutagenesis.70 –75 The methods arecomplementary. The study of multiple clones isolated at different time points (quasispeciesanalysis) is useful to understand the dynamics of viral populations and the emergence ofresistance during therapy.54,56,68,69,91 It helps characterize the in vivo replication fitness ofthe variants in the presence of the drug(s). Improvement and standardization of these methods,however, is needed before their widespread application. Conclusions
The virologic definition of HBV treatment effect, efficacy, and end points allows investigatorsand clinical practitioners to define treatment responses and failures. The currently availableassays for HBV-DNA quantification (in particular those based on real-time PCR), serologicassays for HBeAg and anti-HBe antibodies, and molecular assays for the identification ofgenotypic resistance to HBV drugs can be used confidently to monitor treatment responses anddiagnose primary and secondary treatment failures and their causes in HBV clinical trials andclinical practice. The utility of additional virologic parameters, such as the HBV genotype,quantitative HBsAg, or intrahepatic cccDNA remains to be established. These parametersshould be assessed in clinical trials when possible (but not all clinical trials allow for sequential
liver biopsy specimens for cccDNA assessment).
Diagnosis and monitoring of HBV treatment failure in clinical trials of new anti-HBV drugsinvolves 5 successive steps. First, primary or secondary treatment failure should be diagnosedby means of HBV-DNA measurement according to the earlier-described definitions. Second,nonviral causes of treatment failure should be sought. In this respect, drug dosages areparticularly useful to show poor adherence to therapy, an unusual cause of treatment failure inclinical trials, but the principal one in clinical practice. Third, selection of resistant variantsshould be shown by means of sequencing assays. Fourth, to differentiate true resistancemutations from polymorphisms and to assess the level of resistance to the drug conferred bythe identified mutations, the mutant HBV should be tested for estimation of the IC50 andIC90 against that particular (and other) agent in phenotypic assays, ideally both in and out ofthe context of the full-length patient’s sequence. Fifth, the dynamics of sensitive and resistantviral populations during treatment should be characterized by means of quasispecies analysesat serial time points. In clinical practice, steps 1–3 generally are sufficient to diagnose viral
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
resistance when it is caused by known mutations. When treatment failure is diagnosed andmutations unknown to be associated with resistance to the specific drug are identified, steps 4
and 5 are necessary to confirm that the mutation is indeed responsible for treatment failure.
Standardization of HBV clinical trial reports is now needed based on these definitions and endpoints. Acknowledgements
Supported in part by the VIRGIL European Network of Excellence on Antiviral Drug Resistance, by a grant (LSHM-CT-2004-503359) from the Priority 1 “Life Sciences, Genomics and Biotechnology for Health” program in the 6thFramework Program of the European Union, and by the French National Agency for AIDS and Viral HepatitisResearch (ANRS) (J.M.P. and F.Z.); in part by the Intramural Research Program of the National Institute of Diabetesand Digestive and Kidney Diseases, National Institutes of Health (T.J.L.); and in part by the Extramural ResearchProgram of the National Institutes of Health (RO-1: AIO60449 to S.L.). Abbreviations used in this paper References
1. Lee WM. Hepatitis B virus infection. N Engl J Med 1997;337:1733–1745. [PubMed: 9392700]
2. Ganem D, Prince AM. Hepatitis B virus infection—natural history and clinical consequences. N Engl
J Med 2004;350:1118–1129. [PubMed: 15014185]
3. Zoulim F. Mechanism of viral persistence and resistance to nucleoside and nucleotide analogs in
chronic hepatitis B virus infection. Antiviral Res 2004;64:1–15. [PubMed: 15451174]
4. Shaw T, Bartholomeusz A, Locarnini S. HBV drug resistance: mechanisms, detection and
interpretation. J Hepatol 2006;44:593–606. [PubMed: 16455151]
5. Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced
liver disease. N Engl J Med 2004;351:1521–1531. [PubMed: 15470215]
6. Perrillo RP, Schiff ER, Davis GL, et al. A randomized, controlled trial of interferon alfa-2b alone and
after prednisone withdrawal for the treatment of chronic hepatitis B. N Engl J Med 1990;323:295–301. [PubMed: 2195346]
7. Manesis EK, Hadziyannis SJ. Interferon alpha treatment and retreatment of hepatitis B e antigen-
negative chronic hepatitis B. Gastroenterology 2001;121:101–109. [PubMed: 11438498]
8. Lai CL, Chien RN, Leung NW, et al. A one-year trial of lamivudine for chronic hepatitis B. N Engl J
9. Dienstag JL, Schiff ER, Wright TL, et al. Lamivudine as initial treatment for chronic hepatitis B in the
United States. N Engl J Med 1999;341:1256–1263. [PubMed: 10528035]
10. Leung NW, Lai CL, Chang TT, et al. Extended lamivudine treatment in patients with chronic hepatitis
B enhances hepatitis B e antigen seroconversion rates: results after 3 years of therapy. Hepatology2001;33:1527–1532. [PubMed: 11391543]
11. Cooksley WG, Piratvisuth T, Lee SD, et al. Peginterferon alpha-2a (40 kDa): an advance in the
treatment of hepatitis B e antigen-positive chronic hepatitis B. J Viral Hepat 2003;10:298–305. [PubMed: 12823597]
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
12. Lau GK, Piratvisuth T, Luo KX, et al. Peginterferon alfa-2a, lamivudine, and the combination for
HBeAg-positive chronic hepatitis B. N Engl J Med 2005;352:2682–2695. [PubMed: 15987917]
13. Marcellin P, Lau GK, Bonino F, et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in
combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med 2004;351:1206–1217. [PubMed: 15371578]
14. Lai CL, Gane E, Hsu CW, et al. Two-year results from the GLOBE trial in patients with hepatitis B:
greater clinical and antiviral efficacy for telbivudine (LdT) versus lamivudine. Hepatology 2006;44(Suppl 1):222A.
15. Chang TT, Gish RG, de Man R, et al. A comparison of entecavir and lamivudine for HBeAg-positive
chronic hepatitis B. N Engl J Med 2006;354:1001–1010. [PubMed: 16525137]
16. Chang TT, Gish RJ, de Man RA, et al. Entecavir is superior to lamivudine for the treatment of HBeAg-
positive chronic hepatitis B: results of phase III study ETV-022 in nucleoside-naive patients. Hepatology 2004;40(Suppl 1):193A.
17. Marcellin P, Chang TT, Lim SG, et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-
positive chronic hepatitis B. N Engl J Med 2003;348:808–816. [PubMed: 12606735]
18. Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Long-term therapy with adefovir dipivoxil for
HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology 2006;131:1743–1751. [PubMed: 17087951]
19. Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Adefovir dipivoxil for the treatment of hepatitis
B e antigen-negative chronic hepatitis B. N Engl J Med 2003;348:800–807. [PubMed: 12606734]
20. Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum
hepatitis B virus DNA level. JAMA 2006;295:65–73. [PubMed: 16391218]
21. Iloeje UH, Yang HI, Su J, et al. Predicting cirrhosis risk based on the level of circulating hepatitis B
viral load. Gastroenterology 2006;130:678–686. [PubMed: 16530509]
22. Yu MW, Yeh SH, Chen PJ, et al. Hepatitis B virus genotype and DNA level and hepatocellular
carcinoma: a prospective study in men. J Natl Cancer Inst 2005;97:265–272. [PubMed: 15713961]
23. Wong DK, Cheung AM, O’Rourke K, et al. Effect of alpha-interferon treatment in patients with
hepatitis B e antigen-positive chronic hepatitis B. A meta-analysis Ann Intern Med 1993;119:312–323.
24. Mommeja-Marin H, Mondou E, Blum MR, et al. Serum HBV DNA as a marker of efficacy during
therapy for chronic HBV infection: analysis and review of the literature. Hepatology 2003;37:1309–1319. [PubMed: 12774009]
25. Dienstag JL, Goldin RD, Heathcote EJ, et al. Histological outcome during long-term lamivudine
therapy. Gastroenterology 2003;124:105–117. [PubMed: 12512035]
26. Colonno RJ, Rose RE, Pokornowski K, et al. Assessment at three years shows high barrier to resistance
is maintained in entecavir-treated nucleoside naive patients while resistance emergence increasesover time in lamivudine refractory patients. Hepatology 2006;44(Suppl 2):229A.
27. Di Bisceglie A, Lai CL, Gane E, et al. Telbivudine GLOBE trial: maximal early HBV suppression is
predictive of optimal two-year efficacy in nucleoside-treated hepatitis B patients. Hepatology
28. Yuen MF, Sablon E, Hui CK, et al. Factors associated with hepatitis B virus DNA breakthrough in
patients receiving prolonged lamivudine therapy. Hepatology 2001;34:785–791. [PubMed:11584376]
29. Ide T, Kumashiro R, Koga Y, et al. A real-time quantitative polymerase chain reaction method for
hepatitis B virus in patients with chronic hepatitis B treated with lamivudine. Am J Gastroenterol2003;98:2048–2051. [PubMed: 14499786]
30. Werle-Lapostolle B, Bowden S, Locarnini S, et al. Persistence of cccDNA during the natural history
of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology2004;126:1750–1758. [PubMed: 15188170]
31. Sung JJ, Wong ML, Bowden S, et al. Intrahepatic hepatitis B virus covalently closed circular DNA
can be a predictor of sustained response to therapy. Gastroenterology 2005;128:1890–1897. [PubMed: 15940624]
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
32. Wursthorn K, Lutgehetmann M, Dandri M, et al. Peginterferon alpha-2b plus adefovir induce strong
cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology 2006;44:675–684. [PubMed: 16941693]
33. Laras A, Koskinas J, Dimou E, et al. Intrahepatic levels and replicative activity of covalently closed
circular hepatitis B virus DNA in chronically infected patients. Hepatology 2006;44:694–702. [PubMed: 16941694]
34. Zoulim F. Assessment of treatment efficacy in HBV infection and disease. J Hepatol 2006;44:S95–
35. Yuen MF, Wong DK, Sum SS, et al. Effect of lamivudine therapy on the serum covalently closed-
circular (ccc) DNA of chronic hepatitis B infection. Am J Gastroenterol 2005;100:1099–1103. [PubMed: 15842584]
36. Wong DK, Yuen MF, Yuan H, et al. Quantitation of covalently closed circular hepatitis B virus DNA
in chronic hepatitis B patients. Hepatology 2004;40:727–737. [PubMed: 15349913]
37. Zoulim F. New insight on hepatitis B virus persistence from the study of intrahepatic viral cccDNA.
J Hepatol 2005;42:302–308. [PubMed: 15710212]
38. Hui CK, Bowden S, Jackson K, et al. Clinical significance of intrahepatic hepatitis B virus covalently
closed circular DNA in chronic hepatitis B patients who received cytotoxic chemotherapy. Blood2005;105:2616–2617. [PubMed: 15746088]
39. Kidd-Ljunggren K, Miyakawa Y, Kidd AH. Genetic variability in hepatitis B viruses. J Gen Virol
40. Arauz-Ruiz P, Norder H, Robertson BH, et al. Genotype H: a new Amerindian genotype of hepatitis
B virus revealed in Central America. J Gen Virol 2002;83:2059–2073. [PubMed: 12124470]
41. Chu CM, Liaw YF. Genotype C hepatitis B virus infection is associated with a higher risk of
reactivation of hepatitis B and progression to cirrhosis than genotype B: a longitudinal study ofhepatitis B e antigen-positive patients with normal aminotransferase levels at baseline. J Hepatol2005;43:411–417. [PubMed: 16006001]
42. Orito E, Ichida T, Sakugawa H, et al. Geographic distribution of hepatitis B virus (HBV) genotype
in patients with chronic HBV infection in Japan. Hepatology 2001;34:590–594. [PubMed: 11526547]
43. Grandjacques C, Pradat P, Stuyver L, et al. Rapid detection of genotypes and mutations in the pre-
core promoter and the pre-core region of hepatitis B virus genome: correlation with viral persistenceand disease severity. J Hepatol 2000;33:430–439. [PubMed: 11019999]
44. Funk ML, Rosenberg DM, Lok AS. Worldwide epidemiology of HBeAg-negative chronic hepatitis
B and associated precore and core promoter variants. J Viral Hepat 2002;9:52–61. [PubMed:11851903]
45. Livingston SE, Simonetti JP, McMahon BJ, et al. Hepatitis B virus genotypes in Alaska native people
with hepatocellular carcinoma: preponderance of genotype F. J Infect Dis 2007;195:5–11. [PubMed:17152003]
46. Janssen HL, van Zonneveld M, Senturk H, et al. Pegylated interferon alfa-2b alone or in combination
with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet 2005;365:123–
47. Flink HJ, van Zonneveld M, Hansen BE, et al. Treatment with peginterferon alpha-2b for HBeAg-
positive chronic hepatitis B: HBsAg loss is associated with HBV genotype. Am J Gastroenterol2006;101:297–303. [PubMed: 16454834]
48. Nafa S, Ahmed S, Tavan D, et al. Early detection of viral resistance by determination of hepatitis B
virus polymerase mutations in patients treated by lamivudine for chronic hepatitis B. Hepatology2000;32:1078–1088. [PubMed: 11050059]
49. Gintowt AA, Germer JJ, Mitchell PS, et al. Evaluation of the MagNA Pure LC used with the Trugene
HBV Genotyping Kit. J Clin Virol 2005;34:155–157. [PubMed: 16023890]
50. Hong SP, Kim NK, Hwang SG, et al. Detection of hepatitis B virus YMDD variants using mass
spectrometric analysis of oligonucleotide fragments. J Hepatol 2004;40:837–844. [PubMed:15094233]
51. Hussain M, Fung S, Libbrecht E, et al. Sensitive line probe assay that simultaneously detects mutations
conveying resistance to lamivudine and adefovir. J Clin Microbiol 2006;44:1094 –1097. [PubMed:16517902]
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
52. Kim HS, Han KH, Ahn SH, et al. Evaluation of methods for monitoring drug resistance in chronic
hepatitis B patients during lamivudine therapy based on mass spectrometry and reverse hybridization. Antivir Ther 2005;10:441–449. [PubMed: 15918335]
53. Osiowy C, Villeneuve JP, Heathcote EJ, et al. Detection of rtN236T and rtA181V/T mutations
associated with resistance to adefovir dipivoxil in samples from patients with chronic hepatitis Bvirus infection by the INNO-LiPA HBV DR line probe assay (version 2). J Clin Microbiol2006;44:1994–1997. [PubMed: 16757589]
54. Pallier C, Rodriguez C, Brillet R, et al. Complex dynamics of hepatitis B virus resistance to adefovir
dipivoxil. J Hepatol 2007;46(Suppl 1):S26.
55. Sertoz RY, Erensoy S, Pas S, et al. Comparison of sequence analysis and INNO-LiPA HBV DR line
probe assay in patients with chronic hepatitis B. J Chemother 2005;17:514–520. [PubMed:16323440]
56. Pallier C, Castera L, Soulier A, et al. Dynamics of hepatitis B virus resistance to lamivudine. J Virol
57. Saldanha J, Gerlich W, Lelie N, et al. An international collaborative study to establish a World Health
Organization international standard for hepatitis B virus DNA nucleic acid amplification techniques. Vox Sang 2001;80:63–71. [PubMed: 11339072]
58. Pawlotsky JM. Hepatitis B virus (HBV) DNA assays (methods and practical use) and viral kinetics.
J Hepatol 2003;39(Suppl 1):S31–S35. [PubMed: 14708675]
59. Pawlotsky JM. Molecular diagnosis of viral hepatitis. Gastroenterology 2002;122:1554–1568.
60. Gordillo RM, Gutierrez J, Casal M. Evaluation of the Cobas Taq-Man 48 real-time PCR system for
quantitation of hepatitis B virus DNA. J Clin Microbiol 2005;43:3504–3507. [PubMed: 16000491]
61. Hochberger S, Althof D, Gallegos de Schrott R, et al. Fully automated quantitation of hepatitis B
virus (HBV) DNA in human plasma by the Cobas AmpliPrep/Cobas TaqMan system. J Clin Virol2006;35:373–380. [PubMed: 16461000]
62. Laperche S, Thibault V, Bouchardeau F, et al. Expertise of laboratories in viral load quantification,
genotyping, and precore mutant determination for hepatitis B virus in a multicenter study. J ClinMicrobiol 2006;44:3600–3607. [PubMed: 17021089]
63. Lole KS, Arankalle VA. Quantitation of hepatitis B virus DNA by real-time PCR using internal
amplification control and dual Taq-Man MGB probes. J Virol Methods 2006;135:83–90. [PubMed:16551481]
64. Ronsin C, Pillet A, Bali C, et al. Evaluation of the Cobas AmpliP-rep-total nucleic acid isolation-
Cobas TaqMan hepatitis B virus (HBV) quantitative test and comparison to the Versant HBV DNA3.0 assay. J Clin Microbiol 2006;44:1390–1399. [PubMed: 16597867]
65. Sum SS, Wong DK, Yuen JC, et al. Comparison of the Cobas TaqMan HBV test with the COBAS
Amplicor monitor test for measurement of hepatitis B virus DNA in serum. J Med Virol 2005;77:486–490. [PubMed: 16254975]
66. Punia P, Cane P, Teo CG, et al. Quantitation of hepatitis B lamivudine resistant mutants by real-time
amplification refractory mutation system PCR. J Hepatol 2004;40:986–992. [PubMed: 15158340]
67. Pawlotsky JM. The concept of hepatitis B virus mutant escape. J Clin Virol 2005;34(Suppl 1):S125–
68. Villet S, Ollivet A, Pichoud C, et al. Stepwise process for the development of entecavir resistance in
a chronic hepatitis B virus infected patient. J Hepatol 2006;46:531–538. [PubMed: 17239478]
69. Villet S, Pichoud C, Villeneuve JP, et al. Selection of a multiple drug-resistant hepatitis B virus strain
in a liver-transplanted patient. Gastroenterology 2006;131:1253–1261. [PubMed: 17030194]
70. Chin R, Shaw T, Torresi J, et al. In vitro susceptibilities of wild-type or drug-resistant hepatitis B
virus to (−)-beta-D-2,6-diaminopurine dioxolane and 2′-fluoro-5-methyl-beta-L-arabinofuranosyluracil. Antimicrob Agents Chemother 2001;45:2495–2501. [PubMed: 11502520]
71. Delaney WET, Edwards R, Colledge D, et al. Cross-resistance testing of antihepadnaviral compounds
using novel recombinant baculoviruses which encode drug-resistant strains of hepatitis B virus. Antimicrob Agents Chemother 2001;45:1705–1713. [PubMed: 11353615]
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
72. Delaney WET, Edwards R, Colledge D, et al. Phenylpropenamide derivatives AT-61 and AT-130
inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob Agents Chemother 2002;46:3057–3060. [PubMed: 12183271]
73. Durantel D, Carrouee-Durantel S, Werle-Lapostolle B, et al. A new strategy for studying in vitro the
drug susceptibility of clinical isolates of human hepatitis B virus. Hepatology 2004;40:855–864. [PubMed: 15382118]
74. Walters KA, Tipples GA, Allen MI, et al. Generation of stable cell lines expressing lamivudine-
resistant hepatitis B virus for antiviral-compound screening. Antimicrob Agents Chemother2003;47:1936–1942. [PubMed: 12760870]
75. Ying C, De Clercq E, Neyts J. Lamivudine, adefovir and tenofovir exhibit long-lasting anti-hepatitis
B virus activity in cell culture. J Viral Hepat 2000;7:79–83. [PubMed: 10718947]
76. Shaw T, Edwards R, Sozzi T, et al. Large variation in the replication efficiencies of HBV mutants:
implications for phenotypic assays for antiviral drug resistance. Hepatology 2006;44(Suppl 1):252A. [PubMed: 16799968]
77. Lai CL, Yuen MF. The natural history and treatment of chronic hepatitis B: a critical evaluation of
standard treatment criteria and endpoints. Ann Intern Med 2007;147:58–61. [PubMed: 17606962]
78. Degertekin B, Lok AS. When to start and stop hepatitis B treatment: can one set of criteria apply to
all patients regardless of age at infection? Ann Intern Med 2007;147:62–64. [PubMed: 17606963]
79. Lok AS, Zoulim F, Locarnini S, et al. Antiviral drug-resistant HBV: standardization of nomenclature
and assays and recommendations for management. Hepatology 2007;46:254–265. [PubMed:17596850]
80. Pichoud C, Seigneres B, Wang Z, et al. Transient selection of a hepatitis B virus polymerase gene
mutant associated with a decreased replication capacity and famciclovir resistance. Hepatology1999;29:230–237. [PubMed: 9862871]
81. Locarnini S, Hatzakis A, Heathcote J, et al. Management of antiviral resistance in patients with chronic
hepatitis B. Antivir Ther 2004;9:679–693. [PubMed: 15535405]
82. Delaney WET, Ray AS, Yang H, et al. Intracellular metabolism and in vitro activity of tenofovir
against hepatitis B virus. Antimicrob Agents Chemother 2006;50:2471–2477. [PubMed: 16801428]
83. Haas DW, Smeaton LM, Shafer RW, et al. Pharmacogenetics of long-term responses to antiretroviral
regimens containing efavirenz and/or nelfinavir: an Adult Aids Clinical Trials Group Study. J InfectDis 2005;192:1931–1942. [PubMed: 16267764]
84. Joerger M, Bosch TM, Doodeman VD, et al. Novel deoxycytidine kinase gene polymorphisms: a
population screening study in Caucasian healthy volunteers. Eur J Clin Pharmacol 2006;62:681–684. [PubMed: 16799820]
85. Richman DD. The impact of drug resistance on the effectiveness of chemotherapy for chronic hepatitis
B. Hepatology 2000;32:866–867. [PubMed: 11003636]
86. Lai CL, Dienstag J, Schiff E, et al. Prevalence and clinical correlates of YMDD variants during
lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis 2003;36:687–696. [PubMed:12627352]
87. Lok AS, Lai CL, Leung N, et al. Long-term safety of lamivudine treatment in patients with chronic
hepatitis B. Gastroenterology 2003;125:1714 –1722. [PubMed: 14724824]
88. Colonno RJ, Rose RE, Baldick CJ, et al. High barrier to resistance results in no emergence of entecavir
resistance in nucleoside-naïve subjects during the first two years of therapy. J Hepatol 2006;44(Suppl2):S182.
89. Tenney DJ, Levine SM, Rose RE, et al. Clinical emergence of entecavir-resistant hepatitis B virus
requires additional substitutions in virus already resistant to lamivudine. Antimicrob AgentsChemother 2004;48:3498 –3507. [PubMed: 15328117]
90. Tenney DJ, Rose RE, Baldick CJ, et al. Two-year entecavir resistance in lamivudine refractory HBV
patients reveals different clinical outcomes depending on the resistance substitutions present. Antimicrob Agents Chemother 2007;51:902–911. [PubMed: 17178796]
91. Cane PA, Mutimer D, Ratcliffe D, et al. Analysis of hepatitis B virus quasispecies changes during
emergence and reversion of lamivudine resistance in liver transplantation. Antivir Ther 1999;4:7–14. [PubMed: 10682123]
Gastroenterology. Author manuscript; available in PMC 2009 May 2. Biography
Jean-Michel Pawlotsky acted as a consultant/advisor for Roche (Basel, Switzerland), Gilead
(Foster City, CA), Bristol-Myers Squibb (Princeton, NJ), Idenix (Cambridge, MA), andNovartis (Basel, Switzerland), and received research support from Roche and Gilead; GeoffreyDusheiko received consultancy fees from Glaxo Smith-Kline (London, UK), Roche, Novartis,Schering Plough (Kenilworth, NJ), Idenix, Gilead Sciences, and Bristol-Myers Squibb, andreceived research support from the same groups; Daryl Lau acted as a consultant/advisor andhas received research grants from Roche, Gilead, Bristol-Myers Squibb, and Novartis; GeorgeLau is a Consultant for Roche and Novartis; Stephen Locarnini has received royalties and is apatent holder for Melbourne Health (Parkville, Victoria, Australia), has received consultingfees from Evivar (Melbourne, Victoria, Australia), Gilead, Pharmasset, and Bristol-MyersSquibb, has received research support from Evivar and Gilead, and has ownership interests inPharmasset (Princeton, NJ); Paul Martin is a consultant and speaker for Bristol-Myers Squibb,Gilead, Idenix, and Roche; Douglas D. Richman is a consultant for Bristol-Myers Squibb,Gilead, Idenix, and Roche; and Fabien Zoulim is a consultant and has received speaker feesfrom Gilead, Bristol-Myers Squibb, Idenix, and Novartis. Gastroenterology. Author manuscript; available in PMC 2009 May 2.
Utility of Markers of Treatment Efficacy, Failure, and Resistance
Clinical trials Clinical practice Research tool aProvided that consensus guidelines are available to guide treatment decisions. bIf liver biopsy specimens are available. Gastroenterology. Author manuscript; available in PMC 2009 May 2.
Monitoring Treatment Efficacy and Failure in Clinical Trials and Clinical Practice
Clinical trials Clinical practice
Baseline and every 1–3 months (every month for a new
Baseline and every 1–3 months (every month for a new
aIf a serum HBV-DNA measurement indicates that the patient may have primary or secondary treatment failure, but there is no increase in serum ALTlevel, a second serum HBV-DNA sample should be assayed for confirmation. Gastroenterology. Author manuscript; available in PMC 2009 May 2.
Demonstration of Resistance in Clinical Trials: Indication of Direct SequenceAnalysis of the HBV Reverse Transcriptase (in Both Treatment-Naive and
At the end of 1 year, and every year irrespective of HBV-DNA level
Every case of secondary treatment failure
Gastroenterology. Author manuscript; available in PMC 2009 May 2.
Demonstration of Resistance in Clinical Practice: Indication of Direct Sequence Analysis of the HBV ReverseTranscriptase (in Both Treatment-Naive and Previously Exposed Patients)
Treatment-naive No information Interrupted treatment Failed on treatment Gastroenterology. Author manuscript; available in PMC 2009 May 2.
Neuroscience Letters 406 (2006) 289–292Effect of acute leg cycling on the soleus H-reflex and modifiedAshworth scale scores in individuals with multiple sclerosisRobert W. Motl , Erin M. Snook, Marcus L. Hinkle, Edward McAuley Department of Kinesiology and Community Health, University of Illinois at Urbana-Champaign, 350 Freer Hall, Urbana, IL 61801, United States Received 21 April 20
 NIH Public Access
NIH Public Access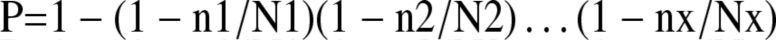 owing to secondary treatment failure, resistance may become a cause of primary treatmentfailure either because of transmission of resistant HBV or because of cross-resistance resulting
from previous therapies. In clinical practice, clinicians likely will be monitoring HBV-DNAlevels and failure to respond to treatment will be the first indication of the emergence ofresistance.
owing to secondary treatment failure, resistance may become a cause of primary treatmentfailure either because of transmission of resistant HBV or because of cross-resistance resulting
from previous therapies. In clinical practice, clinicians likely will be monitoring HBV-DNAlevels and failure to respond to treatment will be the first indication of the emergence ofresistance.