Do you want to buy antibiotics online without prescription? https://buyantibiotics24h.net/ - This is pharmacy online for you!
Neuropsychiatric and cognitive features in autosomal-recessive early parkinsonism due to pink1 mutations
to move her neck in all directions without limitations of
range of motion. Blinking rate is normal and posturaltremor of the hands has also disappeared. REFERENCES
1. Lim EC, Seet RC, Wilder-Smith EP, Ong BK. Dystonia gravida-
rum: a new entity? Mov Disord 2006;21:69 –70.
2. Smith MS, Evatt ML. Movement disorders in pregnancy. Neurol
3. Duane DD. Spasmodic torticollis: clinical and biologic features
David Israeli, MD,3 Oren S. Cohen, MD,1,2
and their implications for focal dystonia. Adv Neurol 1988;50:
Yasaku Hatano, MD,4 Nobutaka Hattori, MD,4 and
4. Cersosimo MG, Bertoti A, Roca CU, Micheli F. Botulinum toxin
in a case of hemimasticatory spasm with severe worsening duringpregnancy. Clin Neuropharmacol 2004;27:6 – 8. 1Department of Neurology, The Sagol Neuroscience Center,
5. Nausieda PA, Koller WC, Weiner WJ, Klawans HL. Chorea in-
Tel-Aviv University, Tel-Aviv, Israel
duced by oral contraceptives. Neurology 1979;29:1605–1609. 2Parkinson’s Disease and Movement Disorders Clinic, The
6. Cogen PH, Zimmerman EA. Ovarian steroid hormones and cere-
Sagol Neuroscience Center, Tel-Aviv University,
bral function. Adv Neurol 1979;26:123–133.
7. Hruska RE, Silbergeld EK. Increased dopamine receptor sensitiv-
ity after estrogen treatment using the rat rotation model. Science
Department of Psychiatry, Chaim Sheba Medical Center,Tel-Aviv University, Tel-Aviv, Israel
8. Miranda M, Cardoso F, Giovannoni G, Church A. Oral contraceptive
4Department of Neurology, Juntendo University School of
induced chorea: another condition associated with anti– basal ganglia
antibodies. J Neurol Neurosurg Psychiatry 2004;75:32–328.
9. Cardoso F. Chorea gravidarum. Arch Neurol 2002;59:868 – 870.
10. Deuschl G, Heinen F, Guschlbauer B, Schneider S, Glocker FX,
Abstract: Autosomal-recessive early-onset Parkinsonism
Lucking CH. Hand tremor in patients with spasmodic torticollis. (AREP) due to PINK1 mutations is characterized by an early-onset, slowly progressive disease, with a good re-
11. Van Hartesveldt C, Joyce JN. Effects of estrogen on the basal
sponse to levodopa. Psychiatric and cognitive disturbances
ganglia. Neurosci Biobehav Rev 1986;10:1–14. associated with AREP have rarely been reported in the
12. Epidemiologic Study of Dystonia in Europe (ESDE) Collaborative
literature. We describe 2 brothers from a Jewish–Iraqi
Group. Sex-related influences on the frequency and age of onset ofprimary dystonia. Neurology 1999;53:1871–1873. consanguineous family with a homozygous PINK1 nonsense
13. Friedman A, Fahn S. Spontaneous remissions in spasmodic torti-
mutation. Both patients presented with anxiety and dyspho-
collis. Neurology 1986;36:398 – 400. ria accompanied by a gait disturbance that developed sub- sequently into a clinical depression. During the course of the disease, both developed drug-induced behavioral dis- turbances of the hedonistic homeostatic dysregulation type and 1 had drug-induced psychosis. The first patient had been diagnosed with mild mental retardation and during the 22 years of disease had further deteriorated; the second developed frontal-type dementia at an early age, 20 years after onset. Their father had a psychiatric disorder but no Parkinsonism. This report expands the phenotypic profile of PINK1-related disease, presenting unique psychiatric and cognitive features as part of the clinical picture. 2007 Movement Disorder Society Key words: Parkinsonism; PINK1; hereditary; cognitive;
The important role of genetics in the etiology of Par-
kinson’s disease (PD) is becoming increasingly recog-
*Correspondence to: Dr. Sharon Hassin-Baer, Department of Neu-
rology and Parkinson’s Disease and Movement Disorders Clinic, SagolNeuroscience Center, Chaim Sheba Medical Center, Tel Hashomer,52621, Israel. E-mail: shassin@post.tau.ac.il
Received 10 May 2006; Revised 28 September 2006; Accepted 3
Published online 26 January 2007 in Wiley InterScience (www. interscience.wiley.com). DOI: 10.1002/mds.21319 Movement Disorders, Vol. 22, No. 4, 2007FEATURES IN AREP DUE TO PINK1 MUTATIONS
nized.1 At present, 10 loci have been identified, repre-
pairment in functions of the frontal network. The patient
senting genomic regions linked to Mendelian forms of
is impulsive with perseverations and marked mental
PD, five of which (PARK 1, 2, 6, 7, and 8) have been
shown to contain gene mutations.2–6 Among the autoso-mal-recessive early-onset parkinsonian (AREP) disor-
Psychiatric Profile
ders, PTEN-induced kinase 1 or PINK1 (PARK6) is the
Antisocial behavior and pathological gambling were
second in prevalence.7 There have only been a handful of
documented several years before presentation. At onset
reports describing the clinical characteristics of patients
of Parkinsonism, he suffered from depressed mood, fa-
with PINK1-associated disease.1,8–12 The known clinical
tigue, palpitations, and dizziness. At that time, the dif-
picture is one of an early-onset (18 –56 years) parkinso-
ferential diagnosis included simple schizophrenia, affec-
nian syndrome, a universally excellent and sustained
tive disorder, and personality disorder. Several trials of
response to levodopa and a protracted course. Psychiatric
antipsychotic drugs caused severe Parkinsonism, which
symptoms, even late in the disease were reported infre-
led to hospitalization. These symptoms improved after
quently.8,9,12 Only 2 cases of patients with cognitive
withdrawal of the drugs. During the past 15 years, he has
impairment were reported, and these conditions devel-
not been hospitalized. At present, he is treated with
oped late in the course of their disease.1,11 Here, we
citalopram and clonazepam with a good effect.
present 2 brothers from a Jewish–Iraqi consanguineousfamily with AREP caused by mutations in PINK1, with
prominent early psychiatric and cognitive features.
The elder brother, presented at age 33 with gait diffi-
culties and anxiety (4 years after his brother). An asym-
metrical parkinsonian syndrome without tremor was ev-
The younger brother, first presented at age 25 with
ident. Spasticity and sleep benefit were absent. There
anxiety and depressive symptoms, along with complaints
was an excellent response to L-dopa. Fluctuations and
of gait difficulties. The prominent psychiatric symptoms
mild dyskinesia evolved after 3 years. After 18 years of
caused a 2-year delay in diagnosis of Parkinsonism.
disease, the most significant motor features are promi-
Finally, however, an asymmetrical parkinsonian syn-
nent motor fluctuations with major gait disturbances dur-
drome without tremor was defined, accompanied by mild
ing off periods. His speech is severely dysarthric and he
lower limb spasticity. Head computed tomography and
has mild dyskinesia. He is, at present, severely disabled
work-up for Wilson’s disease were normal. An excellent
due to dose failures during much of the day, and com-
response to L-dopa and sleep benefit were reported early
monly uses a wheelchair while off. His daily treatment
in the disease course. Motor fluctuations and mild dys-
includes 750 mg of L-dopa divided in six doses, pergol-
kinesia began a year later. During the follow-up period,
the patient remained single and unemployed, despiteonly minimal motor handicap in the first 15 years of
Neurocognitive Profile
disease. At present, 22 years after disease onset, the most
The patient had 9 years of formal education. He had
significant motor features are gait disturbances with se-
been a carpenter and later a postman for several years.
vere freezing of gait, mainly in the off state. His antipar-
Five years ago, he was dismissed from his job as a clerk
kinsonian therapy includes 450 mg of L-dopa divided in
in the post office due to repeated mistakes and negli-
seven doses, entacapone 600 mg, pergolide 0.75 mg,
gence. His wife also complained about his apathy and
amantadine 200 mg, and trihexyphenidyl 3 mg. The
forgetfulness. Executive functions are impaired, and he
patient is adherent with the medication (given borderline
exhibits mental slowness, perseverations, and impulsiv-
Neurocognitive Profile Psychiatric Profile
The patient had 9 years of formal education and had
The patient has a stable marriage and 3 healthy chil-
been diagnosed with mild mental retardation (IQ ϭ 69)
dren. At the age of 33, he had an episode of mixed
during his twenties. Because he never had a stable job or
anxiety and depression along with complaints of “lower
a long-term relationship, it is difficult to estimate the
limb weakness” and was hospitalized in a psychiatric
extent of cognitive deterioration. According to family
department. He suffered from a dysphoric mood, palpi-
members and the decline in his overall functioning, there
tations, and insomnia. He was treated with benzodiaz-
has been a steady deterioration in his cognitive abilities.
epines with marked improvement. During the following
Present neuropsychological work-up shows major im-
years, he remained stable on antidepressant therapy. Four
Movement Disorders, Vol. 22, No. 4, 2007TABLE 1. Neuropsychological profile
Data are presented as Z scores. Case 1: 52 years old, 9 years of education. Case 2: 47 years old, 9 years of education. ND, normative data; MMSE, Mini-Mental State Examination; WAIS, Wechsler Adult Intelligence Scale; AVLT, Auditory Verbal Learning Test;
VOT, Visual Organization Test; WCST, Wisconsin Card Sorting Test.
years ago, severe drug-induced behavioral disturbancesappeared, characterized by hypomanic behavior and hy-persexuality. His behavior was typical of the hedonistichomeostatic dysregulation (HHD)13 with punding behav-ior. His behavior further worsened, and he became dis-inhibited and aggressive, with thoughts of grandeur. Hedid not respond to reduction of dopamine agonist dosageor quetiapine and was hospitalized. His symptoms im-proved after treatment with clozapine. Psychiatric treat-ment today consists of clozapine 50 mg, fluoxetine 20mg, and clonazepam 3 mg. Family History
The family history is negative for Parkinsonism. The
patient’s father suffered from an unspecific psychiatricdisorder, most probably a schizoaffective disorder. Case2 has a healthy twin sister. No other information is
FIG. 1. Square symbols represent men, circles represent women, dia-
monds represent unknown number and gender of siblings, and diagonallines indicate deceased individuals. The pedigree shows that 2 of the 3
Molecular Analysis
siblings are affected (black squares). Both showed a homozygousnonsense mutation in exon 3 (nucleotide 736 C-to-T transition) in
Linkage analysis was negative for Parkin and DJ-1PINK1. The vertically hatched square shows the father who had a
mutations. Both patients are homozygous for a nonsense
psychiatric disorder (information from history only). Movement Disorders, Vol. 22, No. 4, 2007PERMANENT LITHIUM-INDUCED CEREBELLAR TOXICITY
mutation in exon 3 (nucleotide 736 C-to-T transition) in
The father of our patients suffered from an unspecified
PINK1. This mutation was not present in DNA from 50
psychiatric disorder without parkinsonian features, sug-
control Israeli patients of Jewish–Iraqi descent. It prob-
gesting that the PINK1 mutation hemizygous state may
ably leads to premature termination and formation of a
be associated with psychiatric disturbances. Alterna-
truncated protein that lacks 336 amino acids, including a
tively, a different genetic trait may contribute to the
highly conserved protein kinase domain. Genetic analy-
psychiatric disturbances in our patients and their father.
To further explore the possibility of psychiatric disordersattributed to the hemizygous mutation state, family mem-
DISCUSSION
bers of PD patients due to PINK1 mutations should be
We present two brothers from a consanguineous
family with AREP due to a homozygous mutation in
This report expands the phenotypic profile of PINK1-
PINK1. Both patients presented with psychiatric com-
related Parkinsonism, presenting unique psychiatric and
plaints accompanied by a gait disturbance. The psy-
cognitive features as part of the core clinical features of
chiatric features were the major cause of disability in
this disease. Variability in the phenotype of PINK1 mu-
both patients. Both presented with anxiety and dys-
tations highlights the overlap of clinical features in the
phoria developing subsequently into depression. Psy-
three known recessive forms of young-onset Parkinson-
chiatric disturbances were apparent before introduc-
ism, stressing the need for genetic testing for accurate
tion of antiparkinsonian therapy. Although the first
patient was diagnosed with mild mental retardation,during 22 years of disease he deteriorated and like his
REFERENCES
brother, developed dementia at an early age. Case 2
1. Albanese A, Valente EM, Romito LM, et al. The PINK1 phenotype
had no history of mental retardation. He developed an
can be indistinguishable from idiopathic Parkinson disease. Neu-
extensive cognitive decline involving damage to the
frontal lobe network. Although a formal neuropsycho-
2. Lewthwaite AJ, Nicholl DJ. Genetics of parkinsonism. Curr Neu-
logical profile was completed only recently (Table 1),
3. Bonifati V. Genetics of Parkinson’s disease. Minerva Med 2005;
a long-standing cognitive deterioration was apparent
over the years as the brothers’ level of functioning
4. McInerney-Leo A, Hadley DW, Gwinn-Hardy K, et al. Genetic
declined and they became increasingly dependent.
testing in Parkinson’s disease. Mov Disord 2005;20:1–10.
This finding was largely due to cognitive and psychi-
5. Lucking CB, Durr A, Bonifati V, et al. Association between
early-onset Parkinson’s disease and mutations in the parkin gene.
atric, rather than motor, disturbances.
French Parkinson’s Disease Genetics Study Group. N Engl J Med
Few studies describe the clinical features of AREP
patients with PINK1 mutations, and the phenotype is still
6. Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1
gene associated with autosomal recessive early-onset parkinson-
being elucidated. Bonifati and colleagues recently de-
scribed the clinical and genetic characteristics of 10
7. Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-
patients of which 5 had concomitant anxiety and/or de-
onset Parkinson’s disease caused by mutations in PINK1. Science
pression.9 Two additional patients in his series developed
8. Hatano Y, Sato K, Elibol B, et al. PARK6-linked autosomal
drug-induced hallucinations. Hatano and associates re-
recessive early-onset parkinsonism in Asian populations. Neurol-
ported 2 other patients with hallucinations, although it
was not mentioned whether these were drug induced.8
9. Bonifati V, Rohe CF, Breedveld GJ, et al. Early-onset parkinson-
ism associated with PINK1 mutations: frequency, genotypes, and
Ibanez and coworkers described 12 patients, 3 of whom
phenotypes. Neurology 2005;65:87–95.
suffered from depression. In 1 patient, depression was
10. Valente EM, Salvi S, Ialongo T, et al. PINK1 mutations are
diagnosed 20 years after parkinsonian symptoms. In the
associated with sporadic early-onset parkinsonism. Ann Neurol
other 2, depression preceded parkinsonian symptoms by
11. Healy DG, Abou-Sleiman PM, Gibson JM, et al. PINK1 (PARK6)
several years; of interest, these patients were sisters, both
associated Parkinson disease in Ireland. Neurology 2004;63:1486 –
had the same homozygous mutation (Q456X).12
Two patients with cognitive impairment were re-
12. Ibanez P, Lesage S, Lohmann E, et al. Mutational analysis of the
PINK1 gene in early-onset parkinsonism in Europe and North
ported. Healy and colleagues11 described a woman of 64
years of age that developed dementia and visual halluci-
13. Pezzella FR, Colosimo C, Vanacore N, et al. Prevalence and
nations 13 years after disease onset. Li and associates14
clinical features of hedonistic homeostatic dysregulation in Parkin-
described a patient with dementia, depression, and hal-
son’s disease. Mov Disord 2005;20:77– 81.
14. Li Y, Tomiyama H, Sato K, et al. Clinicogenetic study of PINK1
lucinations. Apart from these reports, dementia has not
mutations in autosomal recessive early-onset Parkinsonism. Neu-
yet been described in patients with PINK1 mutations. Movement Disorders, Vol. 22, No. 4, 2007
Benefits Checklist Prescription Drug Assistance Programs Partnership for Prescription Assistance (www.pparx.com) Eligibility information is entered and available options are generated. Yes No Reason________________________________________________________________________________________________________________________________________________________________________________
Cotag from Indala—a new generation of patented Hands-Free radio frequency identification (RFID) technology offering unparalleled user convenience and security. Indala introduces a new line of active cards and tags for a variety of applications—the 928 Hands-Free access card and the 911 Hands-Free keyring. Both are designed to complement the entire line of Cotag Hands-Free long-rang
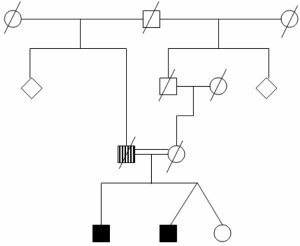 TABLE 1. Neuropsychological profile
TABLE 1. Neuropsychological profile