Do you want to buy antibiotics online without prescription? https://buyantibiotics24h.net/ - This is pharmacy online for you!
Pixuvri, inn-pixantrone
SUMMARY OF PRODUCT CHARACTERISTICS ▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 for how to report adverse reactions. 1. NAME OF THE MEDICINAL PRODUCT
Pixuvri 29 mg powder for concentrate for solution for infusion 2. QUALITATIVE AND QUANTITATIVE COMPOSITION
One vial contains pixantrone dimaleate equivalent to 29 mg pixantrone After reconstitution, each ml of concentrate contains pixantrone dimaleate equivalent to 5.8 mg pixantrone. Excipient with known effect: One vial contains 39 mg sodium. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM
Powder for concentrate for solution for infusion. Dark blue lyophilised powder. 4. CLINICAL PARTICULARS Therapeutic indications
Pixuvri is indicated as monotherapy for the treatment of adult patients with multiply relapsed or refractory aggressive Non-Hodgkin B-cell Lymphomas (NHL). The benefit of pixantrone treatment has not been established in patients when used as fifth line or greater chemotherapy in patients who are refractory to last therapy. 4.2 Posology and method of administration Pixuvri must be administered by physicians who are familiar with the use of antineoplastic agents and have the facilities for regular monitoring of clinical, haematological, and biochemical parameters during and after treatment (see section 6.6). Posology The recommended dose is 50 mg/m2 of pixantrone on days 1, 8, and 15 of each 28-day cycle for up to 6 cycles. Please note: In the EU recommended dose refers to the base of the active substance (pixantrone). Calculation of the individual dose to be administered to a patient must be based on the strength of the reconstituted solution that contains 5.8mg/ml pixantrone and the dose recommendation of 50 mg/m2. In some trials and publications, the recommended dose is based on the salt form (pixantrone dimaleate) However, the dose has to be adjusted before the start of each cycle based on nadir haematologic counts or maximum toxicity from the preceding cycle of therapy. The amount of Pixuvri in milligrams that is to be administered to a patient should be determined on the basis of the patient’s body surface area (BSA). The BSA should be determined using the institutional standard for BSA calculation and should use a weight measured on day 1 of every cycle.
Some caution is advised in obese patients as data on BSA- based dosing is very limited for this group. Dose modification guidelines Dose modification and the timing of subsequent doses should be determined by clinical judgement depending on the degree and duration of myelosuppression. For subsequent courses, the prior dose can usually be repeated if white blood cell and platelet counts have returned to acceptable levels. If on day 1 of any cycle the Absolute Neutrophil Count (ANC) is < 1.0 x 109/l or platelet count is < 75 x 109/l it is recommended to delay treatment until ANC recovers to ≥ 1.0 x 109/l and platelet
count to ≥ 75 x 109/l. Table 1 and Table 2 are recommended as guides to dosage adjustments for days 8 and 15 of the 28- day cycles.
Dose modifications for hematologic toxicity on days 8 and 15 of any cycle Grade Platelet count ANC count Dose modification
Delay treatment until recovery to platelet count
≥ 50 x 109/l and ANC** ≥ 1.0 x 109/l. Delay treatment until recovery to platelet count
≥ 50 x 109/l and ANC** ≥ 1.0 x 109/l. Reduce the dose by 20%.
Treatment modifications for non-hematologic toxicities Toxicity Modification
Any grade 3 or 4 drug-related non cardiac
Delay treatment until recovery to grade 1.
Reduce the dose by 20%. Delay treatment and monitor until
recovery.Consider discontinuation for persistent
decline in LVEF** of ≥ 15% of baseline value.
* NYHA: New York Heart Association ** LVEF: Left Ventricular Ejection Fraction Special populations Paediatric population The safety and efficacy of Pixuvri in children aged < 18 years has not yet been established. No data are available. Elderly patients
No specific dose adjustment is required in elderly patients (aged ≥ 65 years). Patients with impaired renal function The safety and efficacy of Pixuvri has not been established in patients with impaired renal function. Patients with serum creatinine > 1.5 x Upper Limit of the Normal range (ULN) were excluded from the randomised study. Thus, Pixuvri should be used with caution in patients with renal impairment. Patients with impaired hepatic function The safety and efficacy of Pixuvri in patients with impaired hepatic function has not been established. Pixuvri should be used with caution in patients with mild or moderate liver impairment.
Pixuvri is not recommended for use in patients with severe excretory hepatic impairment, (see section 4.3). Patients with poor performance status There is currently no information on the safety and efficacy of patients with poor performance status (ECOG > 2). Caution should be exercised when treating such patients.
Method of administration Pixuvri is for intravenous use only. The safety of intrathecal use has not been established. Pixuvri is intended for administration as a slow intravenous infusion using an in-line filter (over a minimum of 60 minutes) only after reconstitution with 5 ml sodium chloride 9 mg/ml (0.9%) solution for injection and after further dilution with sodium chloride 9 mg/ml (0.9%) solution for injection to a final volume of 250 ml. For instructions on reconstitution and dilution of the medicinal product before administration, see section 6.6. 4.3 Contraindications
- Hypersensitivity to pixantrone dimaleate, or to any of the excipients listed in section 6.1 - Immunisation with live virus vaccines - Profound bone marrow suppression - Severe abnormal hepatic function. 4.4 Special warnings and precautions for use
All initial treatment with Pixuvri should be preceded by a careful baseline assessment of blood counts, serum levels of total bilirubin, serum levels of total creatinine, and cardiac function as measured by left ventricular ejection fraction (LVEF). Myelosuppression Severe myelosuppression may occur. Patients treated with Pixuvri are likely to experience myelosuppression (neutropenia, leukopenia, anaemia, thrombocytopenia, and lymphopenia) with the predominant manifestation being neutropenia. With the recommended dose and schedule, neutropenia is usually transient, reaching its nadir on days 15-22 following administration on days 1, 8, and 15 with recovery usually occurring by day 28. Careful monitoring of blood counts is required, including leukocyte, red blood cells, platelet, and absolute neutrophil counts. Recombinant hematopoietic growth factors may be used according to institutional or European Society for Medical Oncology (ESMO) guidelines. The dose modifications should be considered (see section 4.2). Cardiotoxicity Changes in cardiac function including decreased LVEF or fatal congestive heart failure (CHF) may occur during or after treatment with Pixuvri. Active or dormant cardiovascular disease, prior therapy with anthracyclines or anthracenediones, prior or concurrent radiotherapy to the mediastinal area, or concurrent use of other cardiotoxic medicinal products may increase the risk of cardiac toxicity. Cardiac toxicity with Pixuvri may occur whether or not cardiac risk factors are present. Patients with cardiac disease or risk factors such as a baseline LVEF value of < 45% by multigated radionuclide (MUGA) scan, clinically significant cardiovascular abnormalities (equal to New York Heart Association [NYHA] grade 3 or 4), myocardial infarction within the last 6 months, severe arrhythmia, uncontrolled hypertension, uncontrolled angina, or prior cumulative doses of
doxorubicin or equivalent exceeding 450 mg/m2 should receive careful risk versus benefit consideration before receiving treatment with Pixuvri. Cardiac function should be monitored before initiation of treatment with Pixuvri and periodically thereafter. If cardiac toxicity is demonstrated during treatment, the risk versus benefit of continued therapy with Pixuvri must be evaluated. Secondary malignancy The occurrence of secondary acute myeloid leukaemia (AML) or myelodysplastic syndrome (MDS) is a well described complication of chemotherapy regimens containing anthracyclines and other topoisomerase II inhibitors. Infection Infections, including pneumonia, cellulitis, bronchitis, and sepsis have been reported during clinical trials (see section 4.8). Infections have been associated with hospitalisation, septic shock, and death. Patients with neutropenia are more susceptible to infections, although, in the clinical studies there was no increased incidence of atypical, difficult-to-treat infections, such as systemic mycotic infections or infections with opportunistic organisms such as Pneumocystis jiroveci. Pixuvri should not be administered to patients with an active, severe infection or in patients with a history of recurring or chronic infections or with underlying conditions which may further predispose them to serious infection. Tumour lysis syndrome Pixantrone may induce hyperuricaemia as a consequence of the extensive purine catabolism that accompanies drug-induced rapid lysis of neoplastic cells (tumour lysis syndrome) and can lead to electrolyte imbalances, which can result in kidney damage. Blood uric acid levels, potassium, calcium phosphate, and creatinine should be evaluated after treatment in patients at high risk for tumour lysis (elevated LDH, high tumour volume, high baseline uric acid or serum phosphate levels). Hydration, urine alkalinisation, and prophylaxis with allopurinol or other agents to prevent hyperuricaemia may minimise potential complications of tumour lysis syndrome. Immunisation Immunisation may be ineffective when given during Pixuvri therapy. Immunisation with live virus vaccines is contraindicated due to the immunosuppression associated with Pixuvri therapy (see section 4.3). Extravasation If extravasation occurs the administration should be stopped immediately and restarted in another vein. The non-vesicant properties of Pixuvri minimise the risk of local reaction following extravasation. Prevention of photosensitivity reactions Photosensitivity is a potential risk based on in vitro and in vivo non-clinical data and no confirmed cases have been reported in the clinical trial program. As a precaution, patients should be advised to follow sun protection strategies, including wearing sun protective clothing and using sunscreen. Since most medicinal product-induced photosensitivity reactions are caused by wavelengths within the UV-A range, sunscreen that strongly absorbs UV-A is recommended. Patients on a sodium restricted diet This medicinal product contains approximately 1000 mg (43 mmol) sodium per dose after dilution. To be taken into consideration by patients on a controlled sodium diet.
Interaction with other medicinal products and other forms of interaction
No drug interactions have been reported in human subjects and no drug-drug interaction studies in humans have been performed. In vitro inhibition studies In vitro studies with the most common human cytochrome P450 isoforms (including CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4) have shown a possible mixed-type inhibition of CYP1A2 and CYP2C8 that may be of clinical relevance. No other significant clinically relevant interactions with CYPP450s were observed. Theophylline: when co-administering the narrow-therapeutic index medicinal product theophylline, which is primarily metabolised by CYP1A2, there is a theoretical concern that this substrate may increase in concentration resulting in theophylline toxicity. Theophylline levels should be carefully monitored in the weeks immediately following initiation of Pixuvri concurrent therapy. Warfarin is partially metabolised by CYP1A2, therefore, a theoretical concern exists with regard to co-administration of this medicinal product and the effect inhibition of its metabolism might have on its intended action. Coagulation parameters, specifically international normalised ratio (INR), should be monitored in the days immediately following the initiation of Pixuvri concurrent therapy. Amitriptyline, haloperidol, clozapine, ondansetron and propranolol aremetabolised by CYP1A2, and therefore, a theoretical concern exists that co-administration of Pixuvri may increase blood levels of this medicinal product. Although a risk to inhibition of pixantrone towards CYP2C8 could not be ascertained, caution should be observed when co-administering substances that are primarily metabolised via CYP2C8, such as repaglinide, rosiglitazone, or paclitaxel e.g. by careful monitoring for side effects.
Based on in vitro studies, pixantrone was found to be a substrate for the membrane transport proteins P-gp/BRCP and OCT1 and agents which inhibit these transporters have the potential to decrease hepatic uptake and excretion efficiency of pixantrone. Blood counts should be closely monitored when co-administered with agents which inhibit such transporters such as cyclosporine A or tacrolimus, commonly used to control chronic graft-versus-host disease, and the anti-HIV agents ritonavir, saquinavir, or nelfinavir. In addition, caution should be taken when pixantrone is continuously co-administered with efflux transport inducers such as rifampicin, carbamazepin and glucocorticoids, as pixantrone excretion might be increased with a consequent decrease of systemic exposure. 4.6 Fertility, pregnancy and lactation
Women of childbearing potential Women of childbearing potential and their partners should be advised to avoid pregnancies. Women and men must use effective contraception during and up to 6 months after treatment. Pregnancy There are no data from the use of pixantrone in pregnant women. Studies in animals have shown reproductive toxicity (see section 5.3). Pixuvri is not recommended during pregnancy and in women of childbearing potential not using contraception.
Lactation It is unknown whether Pixuvri/metabolites are excreted in human milk. A risk to the newborn/infants cannot be excluded. Breast-feeding should be discontinued during treatment with Pixuvri. Fertility After repeated administrations of Pixuvri at doses as low as 0,1 mg/kg/day, a dose-dependent testicular atrophy was detected in the dogs. This effect has not been evaluated in humans. As with other agents in the general class of deoxyribonucleic acid (DNA) damaging agents, Pixuvri may be associated with fertility impairment. Whilst the effect on fertility has not been ascertained, a precaution will be to advise male patients to use contraceptive methods (preferably barrier) during treatment and for a period of 6 months post-treatment to allow new sperm to mature. To avoid the risk of long term infertility, sperm banking should be considered. 4.7 Effects on ability to drive and use machines
It is not known whether Pixuvri has an effect on the ability to drive a car or use machines. 4.8 Undesirable effects
Summary of the safety profile The safety of Pixuvri was evaluated in 407 patients. The most common toxicity is bone marrow suppression, particularly of the neutrophil lineage. Although the incidence of severe marrow suppression with clinical consequences is relatively low, patients have been treated with Pixuvri were closely monitored by frequent blood counts, particularly for neutropenia. The incidence of severe infections was low and opportunistic infections associated with immunocompromise were not seen. Although the occurrence of cardiac toxicity manifested by CHF appears to be lower than that would be expected with related medicinal products such as anthracyclines, monitoring of LVEF either by MUGA scans or ECHO is recommended to assess subclinical cardiotoxicity. Experience with pixantrone is limited to patients with LVEF ≥ 45% with
most patients having values ≥ 50%. Experience administering Pixuvri to patients with more significant cardiac compromise is limited and should only be undertaken in the context of a clinical trial. Other toxicities such as nausea, vomiting, and diarrhoea were generally infrequent, mild, reversible, manageable, and expected in patients treated with cytotoxic agents. Effects on hepatic or renal function were minimal or nonexistent. Tabulated list of adverse reactions Adverse drug reactions (ADR) reported with Pixuvri are from final data from all completed studies. ADRs are listed in Table 3 below by MedDRA system organ class and by frequency: very common
(≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000), not known (cannot be estimated from available data). Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Adverse drug reactions reported related to Pixuvri in completed Pixuvri studies by frequency System Organ Class Frequency Undesirable effect
Neutropenic infection, respiratory tract infection,
infection Bronchitis, candidiasis, cellulitis, herpes zoster,
meningitis, nail infection, oral fungal infection, oral
herpes, pneumonia, salmonella gastroenteritis, septic shock
Adverse drug reactions reported related to Pixuvri in completed Pixuvri studies by frequency System Organ Class Frequency Undesirable effect
Neoplasms benign, malignant and unspecified (incl. cysts and
Neutropenia, leukopenia, lymphopenia, anaemia,
Hypersensitivity to the medicinal product
Hyperuricaemia, hypocalcaemia, hyponatraemia,
Taste disturbances, paraesthesia, headache,
Left ventricular dysfunction, cardiac disorder, cardiac
failure congestive, bundle branch block, tachycardia
Pallor, vein discolouration, hypotension
Pleural effusion, pneumonitis, rhinorrhoea
Stomatitis, diarrhoea, constipation, abdominal pain,
Esophagitis, oral paresthesia, rectal haemorrhage
Night sweats, petechiae, rash macular, skin ulcer
Arthralgia, arthritis, back pain, muscular weakness,
musculoskeletal chest pain, musculoskeletal stiffness, neck pain, pain in extremity
Adverse drug reactions reported related to Pixuvri in completed Pixuvri studies by frequency System Organ Class Frequency Undesirable effect
Fatigue, mucosal inflammation, pyrexia, chest pain,
Chills, injection site coldness, local reaction
Alanine aminotransferase increased, aspartate
aminotransferase increased, blood alkaline phosphatase increased, blood creatinine increased
Bilirubin urine, blood phosphorus increased, blood
urea increased, gamma-glutamyltransferase increased, neutrophil count increased, weight decreased
* ADRs discussed below
Description of selected adverse reactions Hematologic toxicities and complications of neutropenia Hematologic toxicities have been the most frequent toxicity observed but they have, in general, been easily managed with immunostimulants and transfusion support as needed. While grade 3-4 neutropenia occurred in the randomised trial more frequently among Pixuvri recipients, it was uncomplicated in the majority of cases, noncumulative and associated with a low incidence of febrile neutropenia or infections. Importantly, growth factor support was not routinely required and transfusions with red blood cells and platelets were uncommon. (See section 4.4) Cardiac toxicity In the study PIX 301, decreased ejection fraction occurred in 13 patients (19.1%) in the Pixuvri group. In 11 Pixuvri-treated patients, these events were grade 1-2 and in 2 patients they were grade 3; these events were transient and not Pixuvri dose related. Cardiac failure events (MedDRA terms cardiac failure and cardiac failure congestive) occurred in 6 patients (8.8%) treated with Pixuvri (2 patients with grade 1-2, 1 patient with grade 3, and 3 patients with grade 5). Three Pixuvri patients (4.4%) had tachycardia, arrhythmia, sinus tachycardia, or bradycardia. A baseline cardiac evaluation with a MUGA scan or an ECHO is recommended, especially in patients with risk factors for increased cardiac toxicity. Repeated MUGA scan or ECHO determinations of LVEF should be considered in patients with risk factors such as high cumulative exposure to prior anthracyclines or significant pre-existing cardiac disease. (See section 4.4) Other common toxicities Skin discolouration and chromaturia are known related effects of Pixuvri administration due to the colour of the compound (blue). The skin discolouration generally disappears over a few days to weeks as the medicinal product is cleared. Reporting of suspected adverse reactions Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via (the national system listed in Appendix V) Overdose
There have been no reports of overdose with Pixuvri. Single doses of pixantrone up to 158 mg/m2 have been given in dose-escalation clinical trials without evidence of dose-related toxicity. If overdose occurs, supportive management is recommended. 5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties Pharmacotherapeutic group: Antineoplastic agents, anthracyclines, and related substances. ATC code: L01DB11 Mechanism of action The active substance of Pixuvri is pixantrone , a cytotoxic aza-anthracenedione. Unlike approved anthracyclines (doxorubicin and others) and anthracenediones (mitoxantrone), pixantrone is only a weak inhibitor of topoisomerase II. Moreover, unlike anthracyclines or anthracenediones, pixantrone directly alkylates DNA forming stable DNA adducts and cross-strand breaks. Furthermore, because it incorporates a nitrogen heteroatom into the ring structure and does not have ketone groups, pixantrone has less potential for generating reactive oxygen species, binding iron, and forming alcohol metabolites that are felt to cause the cardiac toxicity of anthracyclines. Due to this unique structure, pixantrone produced minimal cardiotoxicity in animal models compared with doxorubicin or mitoxantrone. A comprehensive retrospective population PK/PD analysis of Phase 1 trials and combination regimens (Phase 1/2) demonstrated that progression-free survival and Grade 2-3 neutropenia were related to Pixuvri exposure. Clinical efficacy and safety The safety and efficacy of Pixuvri as single-agent therapy were evaluated in a multicentre, randomised, active controlled trial in patients with relapsed or refractory aggressive NHL after receiving at least two prior therapies (PIX301). This study randomised 140 patients (1:1) to treatment with either Pixuvri or to an investigator chosen single-agent chemotherapy on the comparator arm. Patient demographics and baseline disease characteristics were well balanced between the treatment groups, and no statistically significant differences were noted. For the study overall, patient median age was 59, 61% were male, 64% were Caucasian, 76% had Ann Arbor stage III/IV disease at baseline, 74% had a baseline International Prognostic Index (IPI) score ≥ 2,
and 60% had received ≥ 3 prior chemotherapies. Mantle cell lymphoma patients were not included in the pivotal study. Patients in PIX 301 were required to have been sensitive to prior anthracycline therapy (confirmed or unconfirmed CR or PR). There is limited data in patients previously treated with rituximab (38 patients in the Pixuvri arm and 39 patients in the comparator arm). Tumour response was assessed by a blinded independent central review panel according to the international workshop to standardise response criteria for NHL. Patients treated with Pixuvri showed a significantly higher rate of complete responses and unconfirmed complete responses (CR/CRu), and a higher objective response rate (ORR), compared to the comparator group (see Table 4).
Summary of response per independent assessment panel (ITT population) End-of-Treatment End-of-Study Comparator Comparator
PR) The Fisher exact test was used to compare proportions in the Pixuvri and comparator chemotherapeutic groups. Patients treated with Pixuvri demonstrated 40% improvement in progression-free-survival compared to patients treated with comparator agents with 2.7 months longer median PFS ( hazard ratio (HR)=0.60, logrank p=0.005) (see Figure 1 below). The median overall survival for patients treated with Pixuvri was 2.6 months longer compared to patients treated with comparator (HR=0.79, logrank p=0.25) (see Figure 2 below).
PIX301 Progression-free survival - end of study Pixantrone Comparator Event (PD or death) Median PFS (months) Pixantrone Comparator Agents PIX 301 Overall survival–end of study Pixantrone Comparator Event (death) Median OS (95% CI) Pixantrone Comparator Agents
The results in the rituximab pretreated patients still showed superior treatment benefit with Pixuvri over the comparator for overall response rate (31.6% with Pixuvri versus 17.9% with the comparator) and median progression-free survival (3.3 months with Pixuvri versus 2.5 months with the comparator). However, the benefit of Pixuvri has not been established when used as fifth line or greater in patients refractory to last therapy, and there is very limited data in this group of patients. Paediatric population The European Medicines Agency has waived the obligation to submit the results of studies with Pixuvri in infants from birth to less than 6 months of age on the grounds that NHL does not occur in this specified paediatric subset. The European Medicines Agency has deferred the obligation to submit the results of studies with Pixuvri in patients from 6 months to less than 18 years of age with NHL (see section 4.2 for information on paediatric use). This medicinal product has been authorised under a so-called “conditional approval” scheme. This means that further evidence on this medicinal product is awaited. The European Medicines Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary. 5.2 Pharmacokinetic properties Absorption Following intravenous administration, plasma concentrations of pixantrone reached the maximal concentration at the end of infusion and then declined poly-exponentially. The pharmacokinetics of Pixuvri was dose-independent in the 3 mg/m2 to105 mg/m2 dose range and no substantial differences were observed when the medicinal product was given as a single agent or in combination studies. Average exposures as single agent accounted for:
From an analysis of population PK data, for a target recorded dose of 50 mg/m2 of pixantrone the median 28-day cycle exposure was 6320 ng.hr/ml (90% CI, 5990-6800 ng.hr/ml), for 3 doses / 4 week cycle. Distribution Pixuvri has a large volume of distribution of 25.8 l and is approximately 50% bound to plasma proteins. Biotransformation Acetylated metabolites are the major biotransformation products of pixantrone. However, in vitro, conversion of pixantrone into the acetylated metabolites by either NAT1 or NAT2 was very limited. In human urine, the compound was mainly excreted unchanged, and very small amounts of phase I and phase II acetylated metabolites were found. Therefore, metabolism does not appear to be an important elimination pathway for pixantrone. Acetylated metabolites were pharmacologically inactive and metabolically stable. Elimination Pixantrone has a moderate to high total plasma clearance of 72.7 l/hr and a low renal excretion accounting for less than 10% of the administered dose in 0-24 hours. The terminal half-life ranged from 14.5 to 44.8 hr with a mean of 23.3 ± 8.0 (n=14, CV=34%) and a median of 21.2 hr. Due to the limited contribution of renal clearance, plasma clearance is mainly non-renal. Pixuvri may be metabolised in the liver and/or excreted in the bile. As metabolism appears to be limited, biliary excretion of unchanged pixantrone may be the major elimination pathway. Hepatic clearance approximates the hepatic plasma flow, suggesting a high hepatic extraction ratio and, therefore, efficient parent active substance elimination. Hepatic uptake of pixantrone is possibly mediated by OCT1 active transporters and biliary excretion by P-gp and BCRP. Pixantrone had only a weak or no capability to inhibit P-gp, BCRP, and BSEP transport mechanism in vitro. Pixantrone did inhibit OCT1-mediated metformin transport in vitro, but is not expected to inhibit OTC1 in vivo at clinically relevant concentrations. Pixantrone was a poor inhibitor of OATP1B1 and OATP1B3 uptake transporters in vitro. Linearity/non-linearity Pharmacokinetics of pixantrone proved to be linear in a broad range of doses, from 3 mg/m2 to 105 mg/m2. Pharmacokinetic/pharmacodynamic relationship(s) A relationship between plasma exposure to pixantrone and neutrophil count has been observed.
Preclinical safety data
After a single intravenous administration of Pixuvri at 29 mg/kg and 38 mg/kg, immediate deaths were seen in mice (114 mg/m2, LD10). Decreases in white and red blood cells and alterations in bone marrow, spleen, kidney, and testes were observed. Similar findings were reported in rats, and in dogs at 116 mg/m2. In dogs, tachycardia and electrocardiography (ECG) changes occurred immediately after treatment. In repeated-dose studies in mice, rats, and dogs, the main findings were myelotoxicity, nephrotoxicity (except dogs), and testes damage. In dogs, Pixuvri given at 0.5 to 0.9 mg/kg for six cycles did not cause mortality or severe clinical signs, including ECG or body weight changes. Males were more sensitive to treatment, with respect to reduction in white blood cells and platelet count (reversible) and lymphoid depletion (spleen and thymus), as well as the marked toxicity to reproductive organs, as expected from a cytotoxic agent. Except for a transient increase in exposure in females after the third cycle, there were no marked differences in pharmacokinetic parameters. Males showed, however, slightly higher exposure than females. In dogs, the heart was not affected by treatment, as no ECG changes were seen at different treatment times, nor heart changes were detected at gross- and histopathology. Kidney function and histology were similarly not affected both in 4- and 26-week studies. The cardiotoxic potential of Pixuvri compared with equiactive doses of doxorubicin and mitoxantrone in treatment-naïve and doxorubicin-pre-treated mice was evaluated. Pixantrone dimaleate up to 27 mg/kg given twice a week for 4 weeks did not induce any cardiotoxic effects, while mitoxantrone, as expected, was cardiotoxic at all tested doses (0.6, 1.6, and 1.5 mg/kg). Slight nephropathy was induced by Pixuvri. Minimal cardiotoxicity of Pixuvri was also demonstrated with repeat treatment cycles at the same doses. Genotoxicity studies confirmed the potential for clastogenic effects in mammalian cells in vitro and in vivo. Pixuvri was mutagenic in the Ames test, increased the number of chromosomal aberrations in human lymphocytes, and increased the frequency of micronuclei in vivo. Pixuvri caused maternal and foetal toxicity in rats and rabbits, even at a dose as low as 1.8 mg/kg given on days 9-11 of pregnancy, higher doses resulting in abortions and total embryo resorption. Embryotoxicity was characterised by reduced mean foetal weight, foetal malformations and incomplete or delayed foetal ossification. No long-term animal studies have been performed to establish the carcinogenic potential of Pixuvri. No local tolerance study was conducted. Pixuvri has been shown to cause phototoxic effects on 3T3 cells in vitro. In a colony-forming units study in mice, the myelotoxicity of Pixuvri and mitoxantrone administered at their LD10 (pixantrone dimaleate 38 mg/kg and mitoxantrone 6.1 mg/kg) was similar. 6. PHARMACEUTICAL PARTICULARS List of excipients
Sodium chloride Lactose monohydrate Sodium hydroxide (for pH adjustment) Hydrochloric acid (for pH adjustment)
Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in section 6.6. 6.3 Shelf life
Unopened vial 4 years Reconstituted and diluted solution Chemical and physical in-use stability has been demonstrated for 24 hours at room temperature (15°C to 25°C) and daylight exposure in polyethylene (PE) standard infusion bags. From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2°C to 8°C, unless reconstitution and dilution have taken place in controlled and validated aseptic conditions. 6.4 Special precautions for storage
Store in a refrigerator (2°C to 8°C). Keep the vial in the outer carton in order to protect from light. For storage conditions of the reconstituted and diluted medicinal product, see section 6.3. 6.5 Nature and contents of container
Type I glass vial with grey butyl rubber stopper with aluminium seal and red plastic cap containing 50 mg pixantrone dimaleate equivalent to 29 mg pixantrone. Pack size of 1 vial. 6.6 Special precautions for disposal and other handling
Reconstitution and dilution Before reconstitution, visually check the lyophilised powder for any unusual defects e.g. cracking, melted, or a glassy appearance. Aseptically reconstitute each 29 mg vial with 5 ml of sodium chloride 9 mg/ml (0.9%) solution for injection. The lyophilised powder should completely dissolve in 60 seconds with agitation. This yields a dark blue solution with a pixantrone concentration of 5.8 mg/ml. Aseptically withdraw the volume needed for the required dose (based on 5.8 mg/ml concentration) and transfer to a 250 ml infusion bag of sodium chloride 9 mg/ml (0.9%) solution for injection. The final concentration of pixantrone in the infusion bag should be less than 580 microgram /ml based upon input of reconstituted medicinal product. Compatibility with other diluents has not been determined. After transferring, thoroughly mix the contents of the infusion bag. The mixture should be a clear and dark blue solution. Polyethersulfone 0.2 µm pore size in-line filters should be used during administration of the diluted Pixuvri solution. Pixuvri is a cytotoxic agent. Avoid contact with eyes and skin. Use gloves, masks, and protective eyewear when handling Pixuvri and during decontamination procedures.
Special precautions for disposal Pixuvri is for single use only. Any unused medicinal product or waste material including materials used for reconstitution, dilution, and administration should be disposed of in accordance with local requirements applicable to cytotoxic agents. 7. MARKETING AUTHORISATION HOLDER
CTI Life Sciences Limited Highlands House Basingstoke Road Spencers Wood, Reading Berkshire RG7 1NT United Kingdom 8. MARKETING AUTHORISATION NUMBER(S) DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION DATE OF REVISION OF THE TEXT
Detailed information on this medicinal product is available on the website of the European Medicines Agency
A. MANUFACTURER RESPONSIBLE FOR BATCH RELEASE B. CONDITIONS OR RESTRICTIONS REGARDING SUPPLY AND USE C. OTHER CONDITIONS AND REQUIREMENTS OF THE MARKETING AUTHORISATION D. CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND EFFECTIVE USE OF THE MEDICINAL PRODUCT E. SPECIFIC OBLIGATION TO COMPLETE POST-AUTHORISATION MEASURES FOR THE CONDITIONAL MARKETING AUTHORISATION A. MANUFACTURER RESPONSIBLE FOR BATCH RELEASE
Name and address of the manufacturer(s) responsible for batch release
Catalent UK Packaging Limited Lancaster Way, Wingates Industrial Estate Westhoughton, Bolton Lancashire BL5 3XX
B. CONDITIONS OR RESTRICTIONS REGARDING SUPPLY AND USE
Medicinal product subject to restricted medical prescription (see Annex I: Summary of Product Characteristics, section 4.2).
C. OTHER CONDITIONS AND REQUIREMENTS OF THE MARKETING AUTHORISATION
• Periodic Safety Update Reports
The marketing authorisation holder shall submit the first periodic safety update report for this product within 6 months following authorisation. Subsequently, the marketing authorisation holder shall submit periodic safety update reports for this product in accordance with the requirements set out in the list of Union reference dates (EURD list) provided for under Article 107c(7) of Directive 2001/83/EC and published on the European medicines web-portal. D.CONDITIONS OR RESTRICTIONS WITH REGARD TO THE SAFE AND EFFECTIVE USE OF THE MEDICINAL PRODUCT
• Risk Management Plan (RMP)
The MAH shall perform the required pharmacovigilance activities and interventions detailed in the agreed RMP presented in Module 1.8.2. of the Marketing Authorisation and any agreed subsequent updates of the RMP.
• At the request of the European Medicines Agency
• Whenever the risk management system is modified, especially as the result of new
information being received that lead to a significant change to the benefit/risk profile or as the result of an important (pharmacovigilance or risk minimisation) milestone being reached.
If the dates for submission of a PSUR and the update of a RMP coincide, they can be submitted at the same time.
E. SPECIFIC OBLIGATION TO COMPLETE POST-AUTHORISATION MEASURES FOR THE CONDITIONAL MARKETING AUTHORISATION
This being a conditional marketing authorisation and pursuant to Article 14(7) of Regulation (EC) No 726/2004, the MAH shall complete, within the stated timeframe, the following measures:
To conduct a randomised controlled Phase 3 study (PIX306) of pixantrone-
rituximab vs gemcitabine-rituximab in patients with aggressive B-cell NHL, who failed front line CHOP-R who are not eligible for autologous stem cell transplant (ASCT) (2nd line) or failed ASCT (3rd or 4th line). A clinical study report should be submitted.
Thank you for agreeing to participate in the Hispanic Community Health Study/Study of Latinos HCHS/SOL. Your appointment at the HCHS/SOL Study Clinic has been scheduled for: Time: ___:___ A.M. Please come to 450 4th Ave. Suite 311, Chula Vista, CA . A map and directions are attached. For questions, you may call Johanne Hernandez at 619-205-1923, between 9am to 4pm, Monday through Fri
We are glad you are interested in coming to Republic of Congo, and are excited about the possibility of you coming to help us. We designed this packet of information to assist you in preparing for an adventure in service. Please read it carefully, and refer to it often. It contains important information on: Global Outreach Mission (GOM) is an interdenominational mission organization with headqu
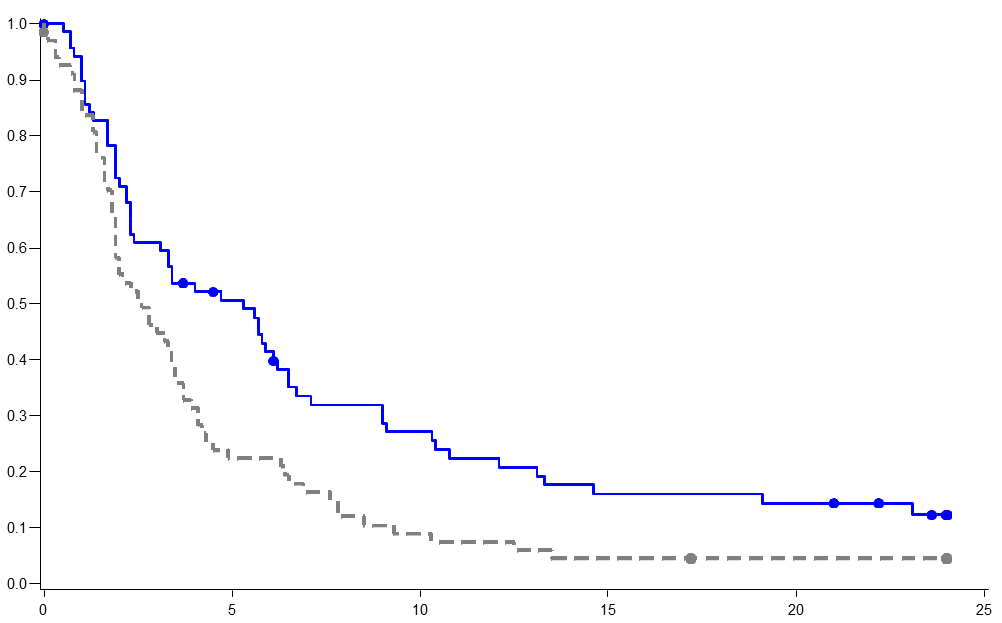 Summary of response per independent assessment panel (ITT population)
Summary of response per independent assessment panel (ITT population) 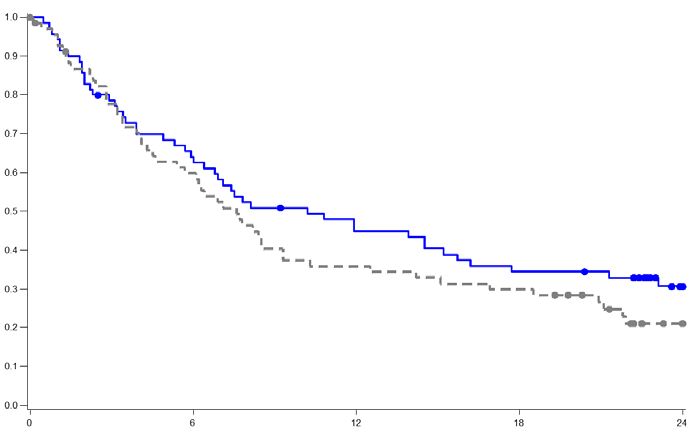 PIX 301 Overall survival–end of study
PIX 301 Overall survival–end of study