Do you want to buy antibiotics online without prescription? https://buyantibiotics24h.net/ - This is pharmacy online for you!
Le-bail-epdic10-proceedings.pdf
Predicted corner-sharing titanium silicates
Université du Maine, Laboratoire des Oxydes et Fluorures, CNRS UMR 6010, Avenue O. Messiaen, 72085 Le Mans Cedex 9, France e-mail: alb@cristal.org Keywords: crystal structure prediction, titanosilicates, inorganic compounds, Monte Carlo Abstract. The prediction of mixed three-dimensional frameworks involving tetrahedra and octahedra sharing exclusively corners is undertaken by applying the Monte Carlo computer program GRINSP (Geometrically Restrained Inorganic Structure Prediction) to titanium silicates. More than 1000 models are selected and stored into the PCOD (Predicted Crystal- lography Open Database). A part of them is identified to real counterparts. The most porous hypothetical structures are presented. The final aim is to build a database of calculated pow- der diffraction patterns for identification by search-match procedures.
As a consequence of the increase in computer power and due to the obvious interest in rely-ing more on planning than on serendipity for chemical synthesis, times are coming for the systematic prediction of the crystal structures of inorganic compounds. A fabulous example consists in the more than 1.500.000 models gathered into the hypothetical zeolites database [1], which should facilitate the identification and structure determination of any new real zeolite by search-match against the calculated powder patterns. The recent publication of the GRINSP (Geometrically Restrained Inorganic Structure Prediction) computer program [2] for the building of N-connected 3D frameworks (N = 3, 4, 5, 6 and binary N/N’ combina-tions) allows for the exploration of single or mixed frameworks of triangles, tetrahedra, square-based pyramids, triangle bipyramids, octahedra or trigonal prisms. Hypothetical GRINSP models built up from corner-sharing TiO6 octahedra and SiO4 tetrahedra are re-ported here. Mixed frameworks, minerals and synthetic compounds, are of great interest, particularly with respect to host-guest chemistry, ion-exchange, adsorption properties, and shape selective catalytic activity. The large class of titanium silicates is represented by more than about 70 minerals, mainly with mixed cation frameworks [3]. They display very excit-ing crystal chemistry and open an attractive outlook to synthesize them and their analogues. Many synthetic homologues of minerals have been reported as well as some new titanium silicates showing open frameworks or bidimensional structures [4,5]. Some compounds ob-tained by thermal decomposition of precursors (hydrates, etc) may not be well enough crys-tallized for a succesful indexing of their powder pattern, so that the key for their structure determination is in the anticipation of their existence. The present predictions by GRINSP
have led to the inclusion of more than 1000 structure-types into the PCOD (Predicted Crys-tallography Open Database) [6].
The knowledge assumed in this study is limited to the M-O, O-O and M-M (M = Si4+, Ti4+) ideal first neighbour distances, and to the exclusive corner-sharing connection mode. GRINSP is a Monte-Carlo (MC) software, applying a pseudo-random number sequence to the heuristic solution of the structural problem. Once a space group (SG) is selected, a first Ti or Si atom (random choice) is placed in a box (with cell parameters relations in agreement with the SG) whose dimensions are selected themselves at random, at a Wyckoff position selected randomly. Then, a second (then a third, etc) Ti or Si atom is placed, if possible. It is checked if the model is not already fulfilling all requirements: one Ti atom should have six Ti or Si (and one Si should have four Ti or Si) first neighbours in the range of M-M distances defined by the user ± 0.4 Å. The fact that distances are given a large tolerance range allows many solutions to be captured which may not correspond to regular polyhedra at this stage. If after some trials, no satisfying model is found, a new first Ti or Si atom will be placed, and so on. For a given set of cell parameters, 300.000 MC events were performed, and at least 20.000 sets of random cell parameters were explored for each SG (230 days of calculation on a single processor running at 2.4 GHz). In this first step, atoms do not move (this is not a simulated annealing approach). The total number of (Ti/Si) atoms placed is not predeter-mined. For that first exploration, the cell parameters were not larger than 16 Å. In a second step the O atoms were added at the midpoints of the Ti -Ti or Ti-Si or Si-Si first neighbours and it was verified by distance and cell improvements (by the MC method as well) that regular TiO6 and SiO4 polyhedra could really be built, i.e. that there was a deep local minima existing close to this previously selected rough arrangement of Ti/Si atoms. The cost function allowing to establish a minimum is based on the verification of the pro-vided ideal distances Si-O (1.61Å), O-O (2.63 Å) and Si-Si (3.07 Å) for SiO4 tetrahedra, and Ti-O (1.95 Å), O-O (2.76 Å) and Ti -Ti (3.80 Å) for TiO6 octahedra. The total R factor is defined by the equation :
where Rn and R0n for n = 1, 2, 3 are defined by the expressions :
where the d0n values for n = 1 to 3 are the ideal first interatomic distances M-O (n = 1), O-O (n = 2) and M-M (n = 3), whereas the dn values are the corresponding observed distances in the structural model for these atom pairs. The terms wn are chosen in order to attribute more weight to the respect of the M-O first distances. When Si-Ti neighbors occur, the ideal dis-tance is estimated as being half the sum of the Si-Si and Ti-Ti distances. Models were re-tained if R < 0.02, they may need further optimizations by using bond valence rules, or en-ergy calculation, however, in most cases the predicted cell parameters differ by less than 2% from the real ones when the real compounds are built up from regular polyhedra, which was the case with dense SiO2 polymorphs or zeolites previously studied by GRINSP [2] and AlF3 phases [7]. During this second step, the atoms are moving, but no jump is allowed because a jump would break the coordinations established at the first step. The change in the cell pa-
rameters from the rough structure candidate to the final model may be quite considerable (up to 30%). During the optimization, the original space group may not be preserved, so that the final structure is always proposed in the P1 space group, presented in a CIF file. An ultimate choice of the real symmetry has to be done by using a program able to detect missing sym-metries, like PLATON [8]. One given model can be identified in different space groups with sligthly different or equal R values. The way GRINSP recognizes a structure-type is by com-paring the coordination sequence (CS) [9] of any model with a list of previously established ones (as well as with the other CS already stored during the current run). That CS, originally developed for zeolites was extended to the N-connected frameworks inside of the GRINSP algorithm. Only one model was retained corresponding to one structure-type, selecting the model with best R value and higher symmetry. GRINSP is available via http://www.cristal.org/grinsp/. The software is free of charge for non-profit organizations and is delivered with the Fortran source code under the GNU Public Licence. Parallelization of the code is in project.
The more than 1000 hypothetical models which were produced exclude structures where edge- or face-sharing polyhedra would occur, and also the structures built up from TiO5 polyhedra (a survey of the TiO5/SiO4 combinations is in progress). A vast majority (~70%) of the hypothetical models proposed by GRINSP corresponds to the general formula [TiSinO(3+2n)]2-. The most numerous models are those with n = 1, 2, 3, 4 and 6, with respec-tively 93, 179, 174, 205 and 158 models satisfying R < 0.02. These models are not electri-cally neutral. For existing, their framework will have to accomodate additional cations or charged molecules. Many known (~50) structures are recognized among the predicted mod-els, some of them are listed in the next paragraph where it is demonstrated that the lack of consideration of an electrical neutrality has limited consequences on the cell parameters. The search by GRINSP is not exhaustive, a new campaign of calculations would allow many of the models to be found again but would also certainly disclose new ones. Moreover, enlarg-ing the cell parameter limit to more than 16 Å would also allow for the building of many new models. The capacity of GRINSP was recently extended to models with a maximum of 192 cations instead of 64 at the time when these calculations were made.
Models corresponding to real structures Identifying the hypothetical models which have a real counterpart is not an easy task. The technique applied here was to make a list of the known titanosilicates or similar materials from the ICSD database and to analyze them one by one, seeing first if they were built up exclusively from corner-sharing TiO6 octahedra and SiO4 tetrahedra, then calculating their CS and comparing to the full list of CS characterizing the hypothetical models. A selected list of frameworks having real titanosilicate counterparts is presented in Table 1. Some have low framework densities (FD : number of Si/Ti atoms in 1000Å3) such as the nenadkevichite structure (FD = 16.4) or the umbite structure adopted by K2TiSi3O9·H2O (FD = 17.2) (figure 1). Considering that GRINSP estimates the cell parameters from the TiO6/SiO4 framework only, without any cation or water molecule (etc) inside of the cages or tunnels, the accuracy of the predictions appears not so bad, as defined by |δ| < 2% (the average absolute value of
the difference in percentage between the observed and predicted cell parameters), with an exception for titanite.
Table 1. Selection of five observed and predicted (italics, second line) [TiSinO(3+2n)]2- frameworks. The PCOD column gives the entry number in the Predicted Crystallography Open Database [6]. Figure 1. Predicted frameworks then identified as corresponding to the known structures nenadkevi-chite (left) and K2TiSi3O9·H2O (right, adopting the umbite mineral structure). Models with the largest porosity The models were classified according to their framework density, providing a list starting at FD = 10.6, corresponding to a porosity P = 70%, close to the smallest known FD values for zeolites. The determination of the pore volumes for the first 10 models in that list was made by using the options SOLV (using r(O) = 1.35 Å, a probe radius of 1.25 Å and a 0.12 Å grid step) of the PLATON computer program which was recently shown to be applicable to stud- ies of microporous inorganic crystals [15]. The porosity P and the volume of pores VP (ac- cording to the definitions in [15]) are presented in Table 2. All of these ten most porous hy- pothetical titanosilicates would behave as zeoates (pores have infinite extension in one direc- tion – becoming a channel – DP = 1, or in 2 or 3 directions – DP = 2 or 3, then the pores form a “channel system”). Not surprisingly, models with the largest porosity have a Si/Ti ratio > 1, however, a Si2Ti3O13 hypothetical compound is the fifth in the list, showing octahedra ar- ranged into HTB (hexagonal tungsten bronze) planes interconnected by tetrahedra. Models denoted A to D, F, H and I in Table 2 have their three-dimensional channels (DP = 3) con-
served with guest diameter > 3 Å whereas models E and J loose it. Model G has DP=1, one of the two channels reducing to isolated pores for larger guests.
Table 2. Pore volumes VP (Å3) are given for the pore dimensionalities DP as derived by SOLV for the ten predicted titanosilicates with smallest framework densities, using a probe radius of 1.25 Å.
Some common building units are observed. Isolated octahedra with 6 corners shared by 6 tetrahedra in two groups of 3 tetrahedra either cyclo-three-connected (case of models A and C), or part of silicate chain with 2 connections (case of models D, F, H and I), or 3 groups of 2 tetrahedra (model J); chains of octahedra trans-connected, the 4 remaining corners satisfied by tetrahedra (case of model B); octahedra organized in HTB sheets interconnected by Si2O7 groups (case of model E); cis-connected chains of either octahedra or trigonal prisms in model G presenting the largest pore aperture with 16-membered ring. Two models are de-picted on figure 2. Figure 2. Predicted cubic frameworks with high porosity (P>60%). Models A (left) and C (right) in the Table 2 notation, both with [Si6TiO15]2- formulation. Extension to isomorphous hypothetical compounds Using GRINS, a satellite program inside of the GRINSP package, the feasibility of isomor- phous compounds was tested. GRINS can read a multiple CIF issued from GRINSP and try a selected cationic/anionic replacement much faster (a few hours for one thousand models)
than if a full prediction was undertaken. Series of zircono-, niobio-, vanadyl-silicates, gallo-, vanadyl-, and titano-phosphates, as well as sulfates, were built starting from the titanium silicates and will be inserted into the PCOD, increasing soon the total number of entries to more than 10.000.
In spite of its limitations, GRINSP appears as an efficient generator of hypothetical crystal structures. Improvements would consist in the consideration of other linkage modes than only by corner-sharing (adding edge and face-sharing) and in the increase of the complexity to combinations of 3 different polyhedra, so as to be able to explore the large domain of the mixed octahedral-pentahedral-tetrahedral framework silicates, for instance. The usefulness of the PCOD will be maximal when powder patterns will be calculated and used by search-match tools for identification purposes. This cannot be done before to fill some frameworks by cations in order to attain electrical neutrality so as to calculate correct diffraction intensi-ties. The target appears to be attainable with automatization (locating holes, filling them with appropriate ions, optimizing according to bond valence rules). Then, some ill-crystallized compounds, with unindexable powder paterns, may well be finally characterized, more or less. Identification will be equivalent to a structure determination, when crystal structure and properties prediction will attain total efficiency, in some expected future.
1. Hypothetical Zeolites Database: http://www.hypotheticalzeolites.net/
2. Le Bail, A., 2005, J. Appl. Cryst. 38, 389-395.
3. Pyatenko, Y.A., Voronkov, A.A. & Pudovkina, Z.V., 1976, The Mineralogical Crystal Chemistry of Titanium (in Russian), (Moscow: Nauka).
4. Rocha, J. & Anderson, M.W., 2000, Eur. J. Inorg. Chem. 5, 801-818.
5. Review in Mineralogy and Geochemistry 57, 2005, Micro- and Mesoporous Min-eral Phases (Editors : Ferraris, G., Merlino, S.).
6. Predicted Crystallography Open Database: http://www.crystallography.net/pcod/
7. Le Bail, A. & Calvayrac, F., 2006, J. Solid State Chem. 179, 3159-3166.
8. Spek, A.L., 2003, J. Appl. Cryst. 36, 7-13.
9. Meier, W.M. & Moeck, H.J., 1979, J. Solid State Chem. 27, 349-355.
10. Hollabaugh, C.L. & Foit, F.F.Jr., 1984, Am. Miner. 69 (1984) 725-732.
11. Rastsvetaeva, R.K., Chukanov, N.V. & Pekov, I.V., 1997, Doklady Akademii Nauk357, 364-367.
12. Dadachov, M.S. & Le Bail, A., 1997, Eur. J. Solid State Inorg. Chem. 34, 381-390.
13. Merlino, S., Pasero, M., Artioli, G. & Khomyakov, A.P., 1994, Am. Miner. 79,
14. Zou, X. & Dadachov, M.S., 2001, J. Solid State Chem. 156, 135-142.
15. Küppers, H., Liebau, F. & Spek, A.L., 2006, J. Appl. Cryst. 39, 338-346.
JOHN F. COOMBS, B.Sc., M.D. 3 WALTER’S LANE, FALLBROOK, ONTARIO Mailing address: P.O. Box 20090, Perth, Ontario, K7H 3M6 Telephone: (613) 267-2523 Fax: (613) 267-6216 HEALTH QUESTIONNAIRE This questionnaire is designed to help you examine some of the many factors affecting your health. It is long and detailed, but the time spent in answering all the questions is well worthw
2007/2008 Lung Health Funding/Clinical Trials RESEARCHER AMOUNT A double-blind, randomized, placebo-controlled, multicenter study to assess the efficacy, safety and tolerability of bosentan in patients with idiopathic Protocol AC-052-321 BUILD 3: Effects of Bosentan on Morbidity and Mortality in Patients With Idiopathic Pulmonary Fibrosis - A Multicenter, Double-Blind, Randomized, Pla
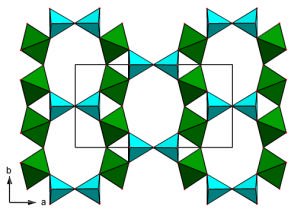
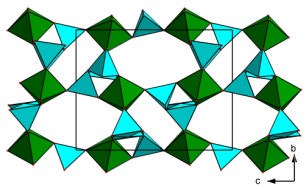 the difference in percentage between the observed and predicted cell parameters), with an exception for titanite.
Table 1. Selection of five observed and predicted (italics, second line) [TiSinO(3+2n)]2- frameworks. The PCOD column gives the entry number in the Predicted Crystallography Open Database [6].
Figure 1. Predicted frameworks then identified as corresponding to the known structures nenadkevi-chite (left) and K2TiSi3O9·H2O (right, adopting the umbite mineral structure).
Models with the largest porosity
the difference in percentage between the observed and predicted cell parameters), with an exception for titanite.
Table 1. Selection of five observed and predicted (italics, second line) [TiSinO(3+2n)]2- frameworks. The PCOD column gives the entry number in the Predicted Crystallography Open Database [6].
Figure 1. Predicted frameworks then identified as corresponding to the known structures nenadkevi-chite (left) and K2TiSi3O9·H2O (right, adopting the umbite mineral structure).
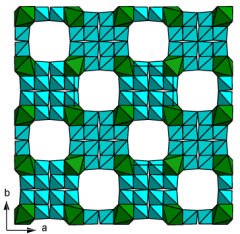
Models with the largest porosity 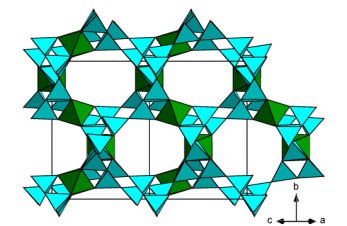
 served with guest diameter > 3 Å whereas models E and J loose it. Model G has DP=1, one of the two channels reducing to isolated pores for larger guests.
Table 2. Pore volumes VP (Å3) are given for the pore dimensionalities DP as derived by SOLV for the ten predicted titanosilicates with smallest framework densities, using a probe radius of 1.25 Å.
Some common building units are observed. Isolated octahedra with 6 corners shared by 6 tetrahedra in two groups of 3 tetrahedra either cyclo-three-connected (case of models A and C), or part of silicate chain with 2 connections (case of models D, F, H and I), or 3 groups of 2 tetrahedra (model J); chains of octahedra trans-connected, the 4 remaining corners satisfied by tetrahedra (case of model B); octahedra organized in HTB sheets interconnected by Si2O7 groups (case of model E); cis-connected chains of either octahedra or trigonal prisms in model G presenting the largest pore aperture with 16-membered ring. Two models are de-picted on figure 2. Figure 2. Predicted cubic frameworks with high porosity (P>60%). Models A (left) and C (right) in the Table 2 notation, both with [Si6TiO15]2- formulation.
Extension to isomorphous hypothetical compounds
served with guest diameter > 3 Å whereas models E and J loose it. Model G has DP=1, one of the two channels reducing to isolated pores for larger guests.
Table 2. Pore volumes VP (Å3) are given for the pore dimensionalities DP as derived by SOLV for the ten predicted titanosilicates with smallest framework densities, using a probe radius of 1.25 Å.
Some common building units are observed. Isolated octahedra with 6 corners shared by 6 tetrahedra in two groups of 3 tetrahedra either cyclo-three-connected (case of models A and C), or part of silicate chain with 2 connections (case of models D, F, H and I), or 3 groups of 2 tetrahedra (model J); chains of octahedra trans-connected, the 4 remaining corners satisfied by tetrahedra (case of model B); octahedra organized in HTB sheets interconnected by Si2O7 groups (case of model E); cis-connected chains of either octahedra or trigonal prisms in model G presenting the largest pore aperture with 16-membered ring. Two models are de-picted on figure 2. Figure 2. Predicted cubic frameworks with high porosity (P>60%). Models A (left) and C (right) in the Table 2 notation, both with [Si6TiO15]2- formulation.
Extension to isomorphous hypothetical compounds